2. SEX, TRAUMA, AND PTSD: WHAT ARE THE DIFFERENCES?
While much of the early research on PTSD involved men with combat-related disorders, population studies have revealed that PTSD is more prevalent in women. In the National Comorbidity Survey,
Kessler et al. (1995) found an overall lifetime prevalence of PTSD of 7.8%, but women were over twice as likely as men to have suffered from the condition (10.4% vs 5.0%;
p < 0.05). Community surveys consistently reveal elevated rates of PTSD among women [e.g., a Swedish study by
Frans et al. (2005)]. Why are most PTSD sufferers female?
Combat exposure accounts for a large proportion of the PTSD seen among men in the United States: the population-attributable fraction is reported to be 27.8% of 12-month PTSD (
Prigerson et al., 2002). The main burden of PTSD in the United States, however, stems not from war or terrorism but from far more common events such as criminal victimization, motor vehicle accidents, and childhood maltreatment (physical, sexual, and emotional) (
Kessler et al., 1995;
Stein, 2002;
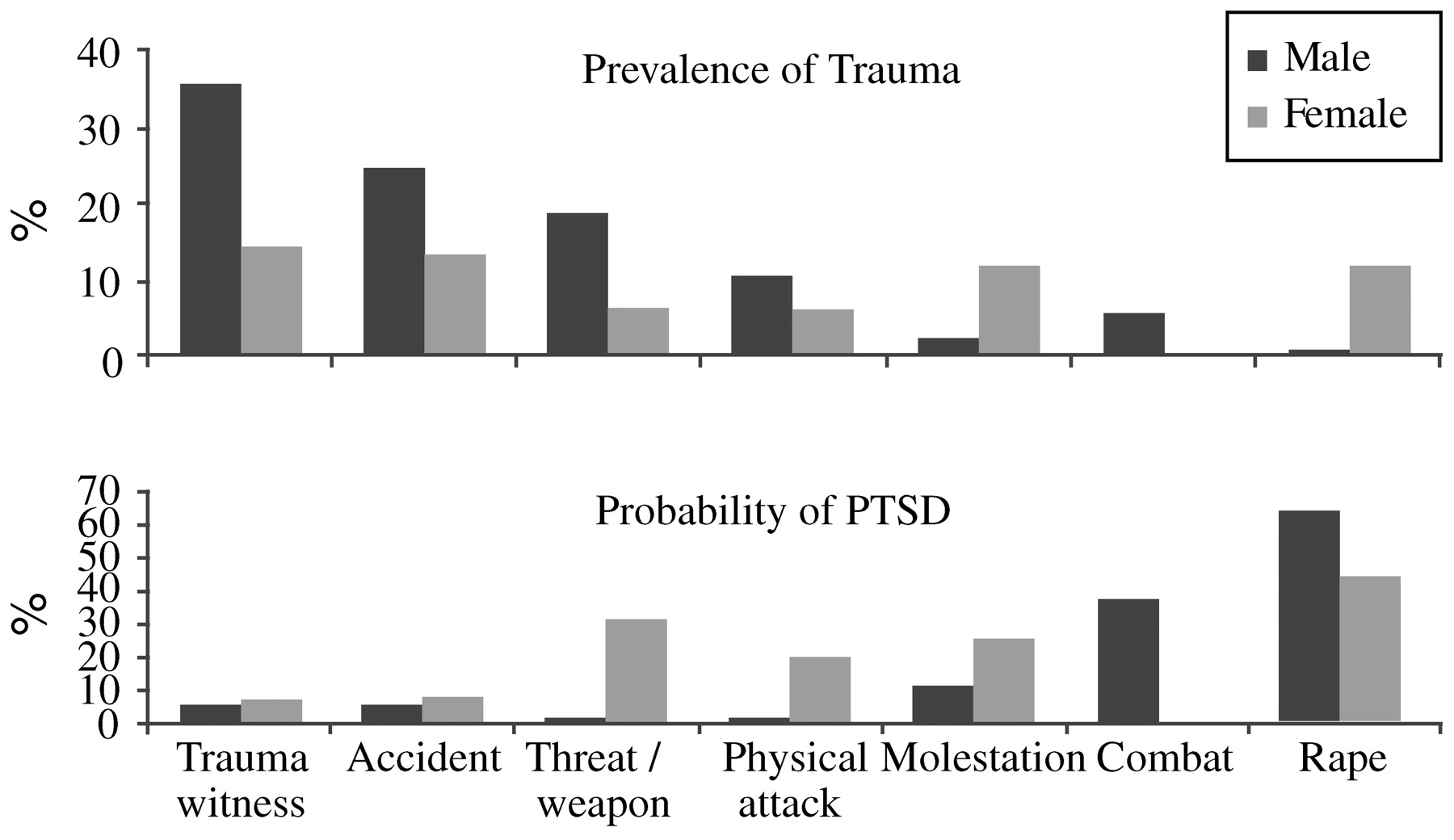
Kessler, 2000). Men and women differ in the types of trauma most frequently encountered; molestation and sexual abuse are more frequent in women, while fights, accidents, and threats involving a weapon (and combat) are more frequent in men. Despite this, even when subjected to the same type of trauma as men, women still have approximately twice the risk of developing PTSD symptoms, and their symptoms are more likely to persist than symptoms among men (
Fig. 1) (
Kessler et al., 1995;
Kessler, 2000;
Breslau et al., 1997;
Breslau and Davis, 1992;
Van Loey et al., 2003;
Holbrook et al., 2002;
Stein et al., 2000;
DeLisi et al., 2003). However, it should be noted that not all studies find this increased susceptibility in women. In a nested case-control analysis of a population-based survey of 30,000 Gulf War era veterans, exposure to either of two kinds of severe trauma—sexual assault and combat—was associated with comparable risk for PTSD in men and women (
Kang et al., 2005). Furthermore, there is preliminary evidence from studies of recent US veterans of war in Iraq and Afghanistan that men and women are at equal risk for PTSD symptoms (
Kang et al., 2005). It may be that gender differences are negligible under circumstances of extreme trauma exposure: this possibility would be compatible with findings from a population-based twin study in which there was an effect of gender on major depression in the presence of lower, but not higher, levels of psychosocial stress (
Kendler et al., 2004). The implications of this observation for mental health prevention efforts remain to be determined.
In one of the very few prospective studies of PTSD,
Perkonigg et al. (2000) followed a German sample of 3021 individuals aged 14–24 and found that predictors of trauma exposure included a pre-existing anxiety disorder (odds ratio (OR) 1.3–3.0) or substance use disorder (OR ∼ 3–7); once trauma had occurred, the strongest predictors of PTSD symptoms were female sex (OR ∼ 2–3) and assault or sexual trauma (OR ∼ 2–4).
Many studies have now demonstrated that female gender is a strong risk factor for the development of PTSD (
Stein, 2002). Though this gender difference is fairly clear from an epidemiologic perspective, the mechanisms for this disparity are uncertain; they may involve both differences in types of trauma exposure and differences in response to trauma. Conceivably, there may be a biological basis to women's apparent vulnerable to certain kinds of traumatic stressors.
Barr et al. (2004) have shown that the serotonin transporter promoter polymorphism (5-HTTLPR) modulates the effect of early adversity in female—but not male—macaque monkeys. They speculate that women with the short (“s”) allele may be more susceptible to the effects of early adversity, a mechanism that explains the increased risk among women for certain stress-related syndromes such as PTSD This work remains to be replicated and extended to humans, but it does pose a testable hypothesis for gender differences in PTSD.
Regarding explanations that focus on differences in type of exposure, violence at the hands of an intimate partner is an epidemic problem that predominantly affects women; it contributes significantly to the burden of PTSD in the female population.
Plichta and Falik (2001) used a nationally representative sample of 2850 American women from the Commonwealth Fund's 1998 Survey of Women's Health to estimate that over 40% of women had experienced some form of violence; 34.6% had experienced intimate partner violence, and 8% had been physically abused by their partners within the previous 12 months. Victims of intimate partner violence often develop psychiatric disorders. In this context,
Bennice et al. (2003) suggested that sexual violence was a greater risk factor than physical violence for the development of PTSD symptoms. In a sample of 44 women who had been victims of intimate partner violence within the last 2 years,
Stein and Kennedy (2001) found that PTSD had a lifetime prevalence of 50%, and 31.8% of PTSD related to violence by intimate partners. Most of the participants in this study had experienced childhood maltreatment, and it was therefore not possible to explore the impact of this factor on PTSD risk. However, other studies have shown that adults with histories of childhood trauma have increased rates of PTSD. Although childhood sexual trauma has been the most intensively studied in this regard, other types of maltreatment—emotional abuse in particular—may be at least as important in explaining gender differences in PTSD. Studying 8667 adult members of an HMO,
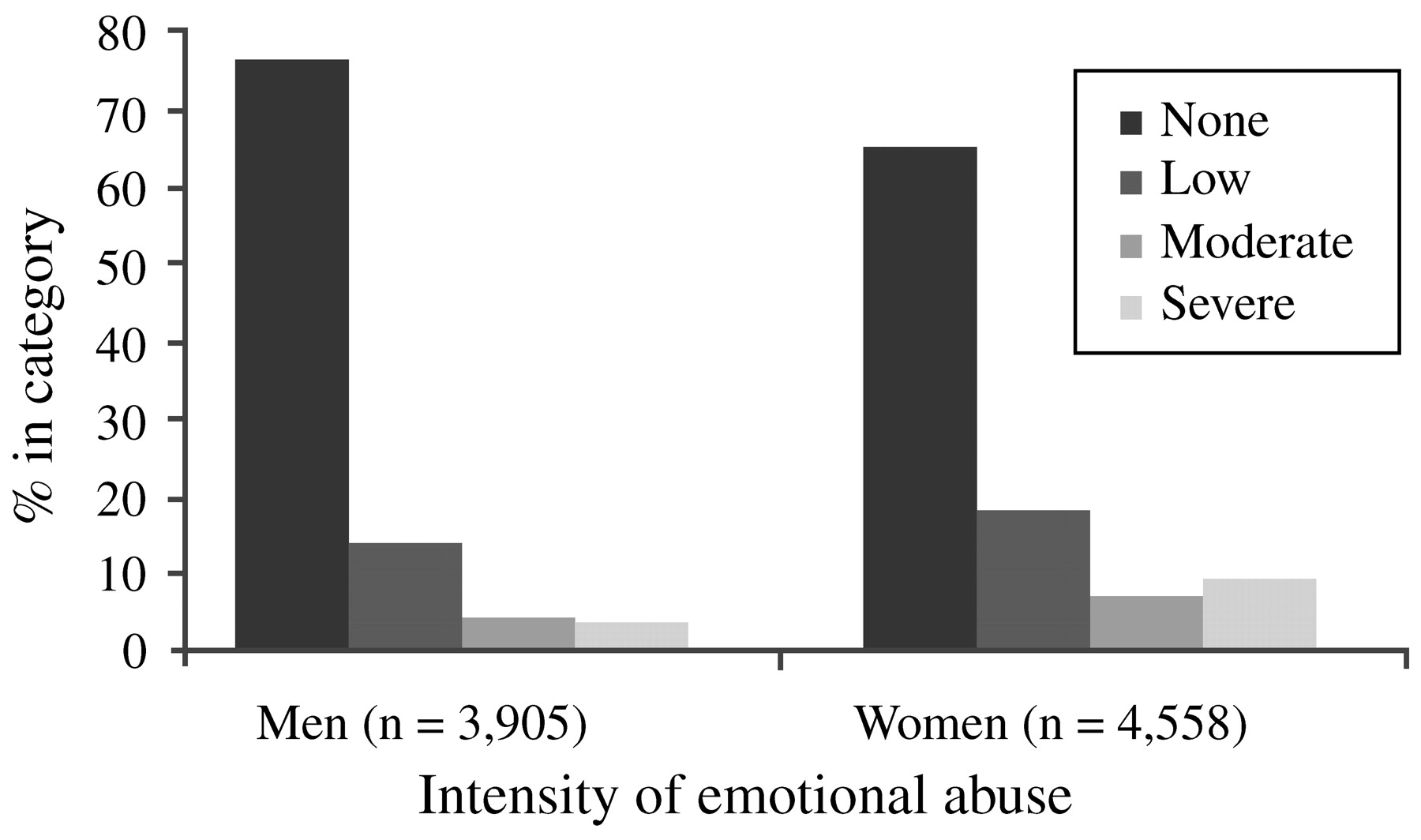
Edwards et al. (2003) found that lower scores on the mental health scale of the SF-36 were associated with more categories of abuse (sexual, physical and emotional abuse, and witnessing of maternal battering), and that both an emotionally abusive family environment and the interaction of such an environment with the different types of maltreatment significantly lowered mental health scores. While men reported significantly more physical abuse, women reported a significantly higher prevalence of sexual abuse as well as moderate or severe emotional abuse in the family environment (
Fig. 2). Thus, differences in types of trauma exposure—possibly relating to sexual trauma (in childhood and adulthood) and intimate partner violence—may explain some of the gender differences in rates of PTSD.
Psychosocial factors such as social support and stigma are also likely to influence gender differences in PTSD following trauma exposure.
Andrews et al. (2003) assessed 118 male and 39 female victims of violent crime for PTSD symptoms as well as for levels of positive support and negative reactions from others. They found that women reported significantly more negative responses from family and friends, and that this factor mediated the relationship between trauma exposure and subsequent PTSD symptoms at 6 months post-exposure. This study serves to remind us that it may be unwarranted to assume that men and women's experience of the same type of trauma will be similar. For example, motor vehicle collisions—even those that are similar in extent of personal injury or vehicular damage—may be experienced very differently by men and women, perhaps in large part because of psychosocial factors (e.g., spousal and/or community support) that mediate the response to trauma. The existence of such factors does not negate the epidemiological fact that women are more at risk for PTSD after many types of trauma exposure, but it may provide another mechanistic explanation for these differences.
The presence of PTSD adversely affects women's health and functioning in many domains (
Dobie et al., 2004;
Ouimette et al., 2004). Walker et al. found that in a sample of 1225 women belonging to an urban HMO, health care costs were doubled among those with high PTSD scores (≥45 on the PTSD Symptom Check-list) (
Walker et al., 2003). Better identification of PTSD in primary care settings might well lead not only to appropriate treatment but also to cost savings.
Do women differ from men in the extent to which they have conditions comorbid with PTSD? In the National Comorbidity Survey,
Kessler et al. (1995) found that, compared with men with PTSD, women with PTSD had lower rates of comorbid alcohol abuse or dependence (27.9% vs 51.9%), drug abuse or dependence (26.9% vs 34.5%), but higher rates of panic disorder (12.6% vs 7.3%) and agoraphobia (22.4% vs 16.1%). However, other data suggest that prior substance abuse may increase the risks of trauma exposure and PTSD, and that childhood maltreatment may increase the risk of substance abuse (
McCauley et al., 1997). Rates of social phobia, major depression, and dysthymia were similar, but a more recent study by
Oquendo et al. (2003) showed that among 156 inpatients with major depression, those with comorbid lifetime PTSD were more likely to have attempted suicide than those without comorbid PTSD (75% vs 54%;
p ≤ 0.01), and among the subgroup with both conditions, the risk of a suicide attempt was higher among women than among men.
Finally, it is possible that there may be gender differences in response to drug treatment for PTSD. However, an early study by
Brady et al. (2000) showing that men responded poorly to treatment with sertraline was not confirmed by later studies using larger sample sizes.
3. RISK AND RESILIENCE FACTORS AFTER DISASTERS AND TERRORISM
Resilience has been addressed since ancient times by authors as diverse as Confucius (“Our greatest glory is not in never falling, but in rising every time we fall”) and Nietzsche (“That which does not kill us can only make us stronger”).
Merriam-Webster's Collegiate Dictionary (
M-W Collegiate Dictionary, 1993) defines resilience as: (1) the capability of a strained body to recover its size and shape after deformation and (2) an ability to recover from or adjust easily to misfortune or change. The metaphor of flexibility and elasticity is also apparent in mental health definitions, which involve the capacity to bounce back, withstand hardship and repair oneself (
Wolin, 1993), or to master cycles of disruption and reintegration (
Flach, 1980). Psychiatric resilience can be defined as resistance to and rapid recovery from psychiatric illness. A wealth of elements that comprise resilience has been proposed in the literature—including active problem-solving, responsibility, self-esteem, independence, well-being, initiative, humor, insight, creativity, and many others. Measuring these concepts and understanding their respective roles presents a formidable challenge.
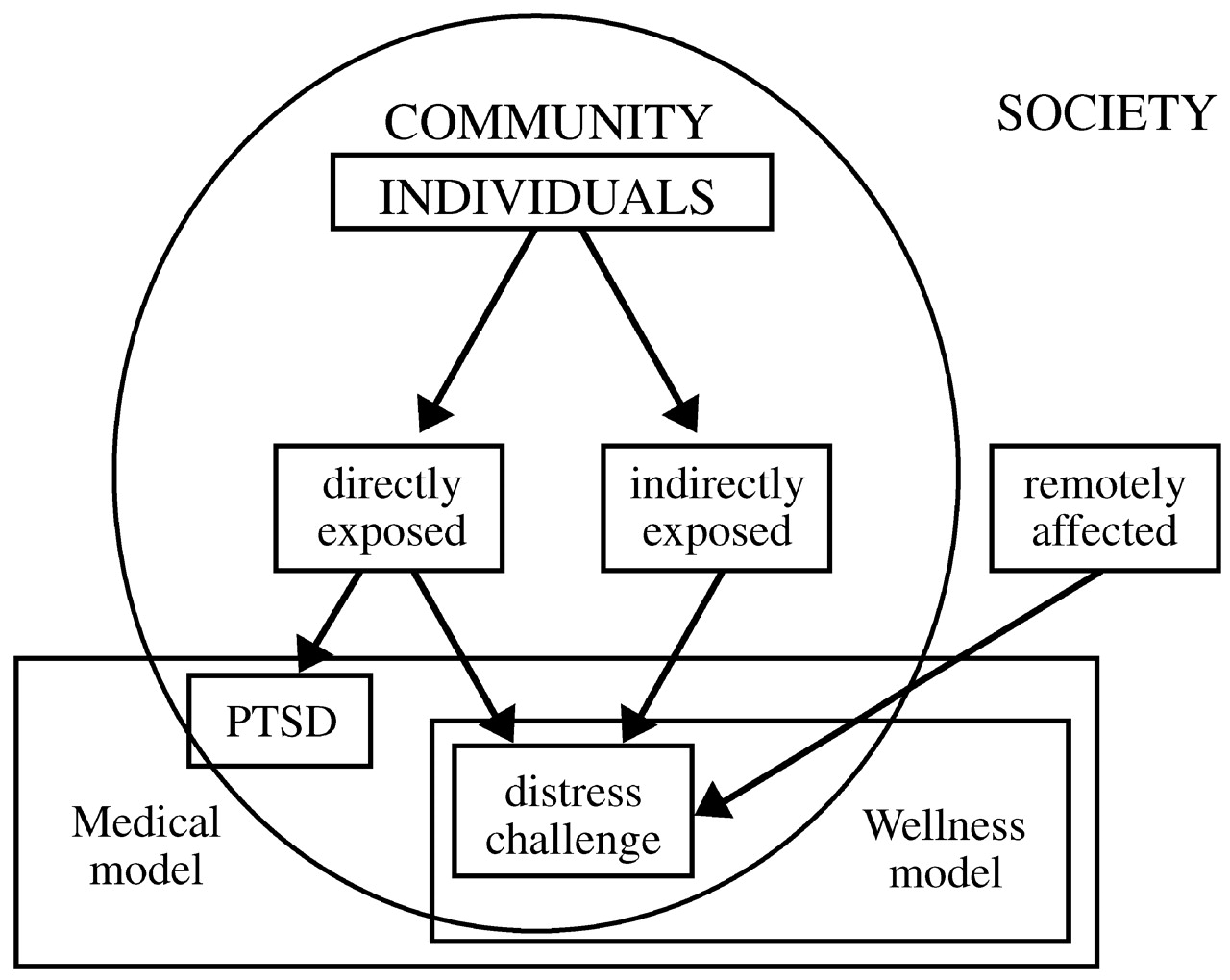
Disasters tend to be random events that expose unselected populations to trauma. Thus, they offer unique opportunities for researchers interested in studying risk and resilience to disentangle the confounding issue of pre-existing risk for exposure to traumatic events. Within a given community, individuals who are highly exposed to a traumatic experience—who are directly in harm's way—will be distressed and challenged by their experience, but only some of them will develop PTSD. A second group of individuals are indirectly exposed to the trauma—they may have lost jobs or had electrical and water utilities cut off, or sustained minor property damage. Finally, if the traumatic event is massive, many individuals outside the particular community may be remotely affected, as much of the American population was affected after September 11, 2001 (
Fig. 3). Each of these groups deserves separate formal intervention. PTSD and other psychiatric disorders are the domain of a medical model that deals with disease and subsumes wellness models that focus on restoring homeostasis. These models need not be in conflict with each other; together they form a comprehensive whole that addresses the effects of disasters, both negative (distress, symptoms, disease) and positive (personal challenge and growth).
Epidemiological studies that attempt to estimate the level of psychopathology in a population after a disaster often neglect important aspects of the PTSD diagnosis. Many popular questionnaires do not determine the sufficiency of exposure to the traumatic event, separate new from pre-existing symptoms, or inquire into the duration of the symptoms or the duration or extent of the resulting disability. Epidemiological estimates of prevalence of PTSD may be inflated as a result.
North et al. (1999) used structured diagnostic interviews to assess 182 adult survivors of the bombing of the Alfred P. Murrah Federal Building in Oklahoma City. Interviewees were selected randomly from the Oklahoma City Health Department's bombing registry. Of these individuals, 87% reported injuries, and 77% had required medical intervention for them. At 6 months after the disaster, even in this highly exposed group, 55% had no psychiatric diagnosis. PTSD was seen in 36% of this population, and 2% of these survivors had PTSD related to traumatic events other than the bombing. Development of PTSD was generally rapid—76% of cases began on the day of the bombing, 94% within the first week, and 98% within the first month. The strongest predictors of PTSD were female sex (45% vs 23%;
p = 0.002) and a pre-disaster history of psychiatric illness (45% vs 26%;
p = 0.009). Over half of those with PTSD had a comorbid psychiatric diagnosis, usually major depression. The second most common psychiatric diagnosis was major depression, seen in 23% of the sample; 78% of those with prior history of major depression had recurrent or persistent major depression at the time of assessment. All cases of substance abuse seen in this sample had developed prior to the bombing, and no new cases were seen.
Individuals with PTSD reported widespread social and occupational dysfunction. For example, 52% of those with PTSD alone and 87% of those with PTSD and a comorbid diagnosis reported some type of functional interference, compared with 27% of those with a non-PTSD diagnosis and 16% of those with no psychiatric diagnosis.
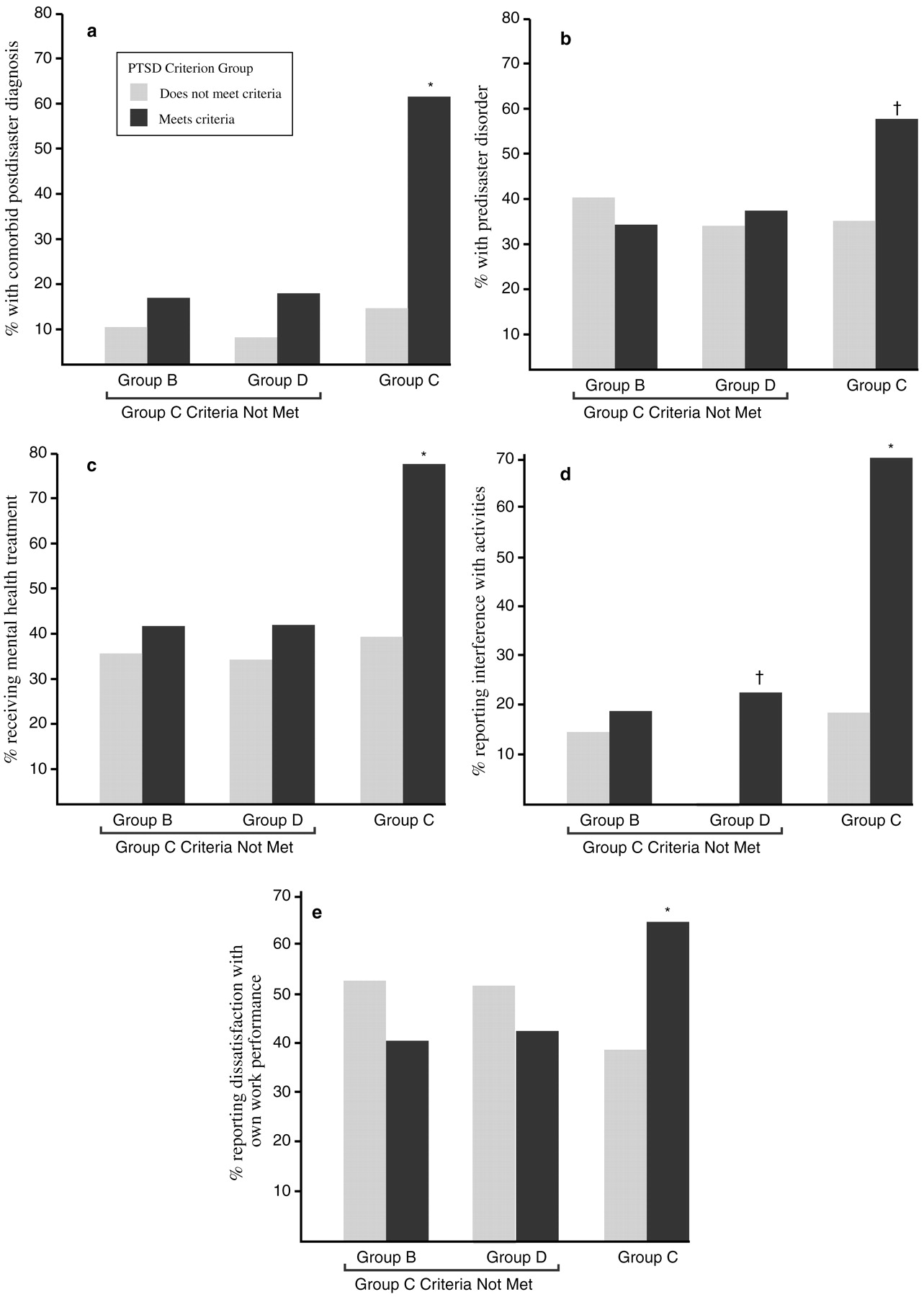
Over the first 6 months after the disaster, 79% of the study participants met the DSM-III-R criterion for group B symptoms (intrusive memories, dreams or nightmares of the event, flashbacks, and being upset by reminders), and 82% met the criterion for group D symptoms (insomnia, difficulty in concentrating, irritability, hypervigilance, and being jumpy or easily startled). However, only 36% met the criteria for group C symptoms (avoidance, psychogenic amnesia, detachment or estrangement, loss of interest, restricted affect, and a sense of shortened future). Of those who met the group C symptom criteria, 94% had a diagnosis of PTSD. The presence of group C symptoms also predicted other diagnoses, the presence of comorbid conditions, interference with activities, dissatisfaction with work performance, and receiving mental health treatment (
Fig. 4).
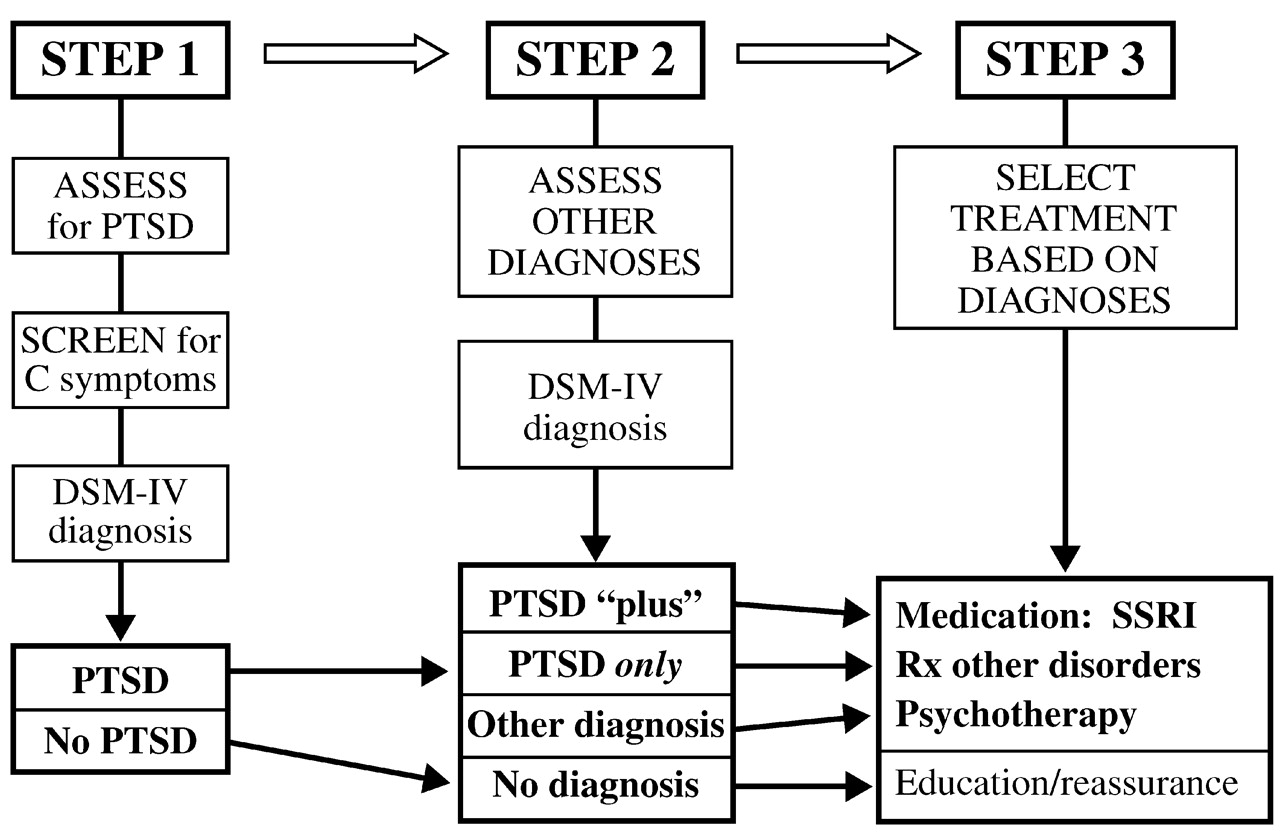
The above results suggest that a simplified algorithm could be constructed for mental health assessment and triage in the aftermath of a disaster. The first step would be to assess for PTSD, the most common diagnosis; if the population at risk is too large for full assessment of everyone, this task may be facilitated by screening for group C criteria, which constitute a marker for the likelihood of PTSD. The second step would be to assess for other diagnoses, whose presence may be critical to selection and outcome of treatment. Finally, treatment would be selected based on the diagnosis (
Fig. 5).
A recent analysis of 160 samples of disaster victims (
Norris et al., 2002) indicated that impairment and adverse outcomes were associated with female gender, ethnic minorities, youthfulness, prior psychiatric problems, secondary stressors, inadequate psychosocial resources, developing rather than developed countries, severity of exposure, and mass violence (e.g., terrorism, shooting sprees) rather than natural or technological disasters.
What about the resilience of those in the larger community—the intended target of demoralization and intimidation by terrorist groups—who are indirectly affected by disasters? While individuals directly exposed to the September 11 disaster have not yet been studied as were the Oklahoma City survivors, numerous studies of indirectly exposed groups have found that 1–2 months after the attacks, 7.5% of a representative sample of the adult Manhattan population reported posttraumatic symptoms (
Galea et al., 2002). A variety of risk factors were identified, including prior stressors, residence near the World Trade Center, loss of possessions or job due to the attacks, the death of a friend or relative during the attack, and low levels of social support. At least two national studies (
Schlenger et al., 2002;
Schuster et al., 2001) have reported an association between extensive viewing of television coverage and substantial stress reactions, although the causal directionalities of this association have not been elucidated. A Pew poll (
Associated Press, 2001) found that a few days after the attacks, 90% of a nationally representative sample found that 7 of 10 reported feeling depressed, half had trouble concentrating, and one third had disturbed sleep. However, a longitudinal study by
Silver et al. (2002) found that the prevalence of posttraumatic stress symptoms related to September 11 among the US population outside New York City declined from 17% at 2 months to 5.8% at 6 months. Not surprisingly, coping strategies assessed shortly after the attacks were the strongest predictors of posttraumatic stress symptoms. Avoidance and numbing symptoms, reflecting inability to cope effectively, were associated with several indicators of problem functioning; these findings were consistent with those of a study of a mass shooting episode in which early coping problems were found to predict PTSD 3 years later (
North et al., 2001).
How can a community's resilience to disasters be increased? Policies and practices that address this question need to be empirically driven, tailoring the interventions to the needs of the individuals being served, rather than the ideologies of service providers. It is essential to recognize and treat the subset of community members with psychiatric illnesses—facilitated by screening for group C symptoms—and to find ways to assist people who have group B and D symptoms (which are nearly ubiquitous among high-exposure groups) without pathologizing them. Mass cognitive reframing could help to correct widespread cognitive distortions and substitute more rational and adaptive thoughts. In this regard, the potential for positive effects of disasters should not be overlooked. Amazingly, many participants in disaster studies have reported favorable outcomes; a review by
McMillen (1999) reported strengthened spiritual or religious beliefs in 15–40% and improvements in interpersonal relationships with family or friends in 10–40%. Finally, a focus on community cohesiveness may also be useful to help people pull together their individual resilience for the good of the greater community.
4. EARLY LIFE TRAUMA AND PTSD
Until the last decade, the hypothesis that early life trauma is associated with an increased risk of adult mood and anxiety disorders was supported largely by anecdotal reports inspired by psychoanalytic concepts of early critical periods of development. Research on the biology of depression and some anxiety disorders has commonly been plagued by the confounding factor of early life stress—the hypercortisolemia and structural brain changes attributed solely to the disorder is likely due, at least in part, to the biological sequelae of early life trauma.
If early trauma is indeed a principal risk factor for later depression and anxiety disorders, one would expect the baseline prevalence of such trauma histories to be very high. However, prevalence data are surprisingly sparse, being derived mainly from small samples or spontaneous reports of trauma from social service departments or hospital emergency rooms. We do know that reported cases constitute a relatively small fraction of all cases, and that although prevalence estimates of childhood abuse and other traumas such as early loss of parents are extremely approximate, they are indeed sufficient to account in part for the high prevalence of depression and anxiety disorders among the general adult population.
In a retrospective study involving 17,337 adult HMO members,
Dube et al. (2001) examined the relationship between adverse childhood experiences and later suicide attempts. In this population, whose mean age was 57 years and which was 54% female, the lifetime prevalence of any suicide attempt was 3.8%, but adverse childhood experiences of any type (emotional, physical, or sexual abuse, substance abuse, mental illness or incarceration of a family member, or parental domestic violence, separation or divorce) increased this risk 2- to 5-fold. Moreover, the greater the number of adverse experiences, the greater the risk of suicide attempts; those with seven or more such experiences had an adjusted odds ratio of 31.1 for a suicide attempt compared with those who had none of these adverse experiences.
McHolm et al. (2003) found consistent results in a community sample of 347 Canadian women aged 15–64 with major depressive disorder: suicidal ideation among them was most strongly associated with a self-reported history of childhood physical abuse (OR 2.77; 95% CI = 1.26–6.12).
The striking results of a study by
McCauley et al. (1997) have also received a great deal of attention, though it has been criticized for the potential confound of retrospective reporting. This cross-sectional survey of 1,931 women from all socioeconomic classes, drawn from four community-based primary care internal medicine practices, found a 22% prevalence of reported childhood or adolescent physical or sexual abuse. Compared with the remainder of the sample, those with childhood, but not adult, abuse histories reported significantly more physical symptoms (mean 6.2 vs 4.0;
p < 0.001), as well as significantly higher Symptom Checklist-22 scores for depression, anxiety, somatization, and interpersonal sensitivity, a 4.7-fold higher prevalence of drug abuse, a 2.2-fold higher alcohol abuse, a 3.7-fold higher history of suicide attempts, and a 3.2-fold higher risk of psychiatric admission to hospital.
More recently,
Safren et al. (2002) found that patients with panic disorder were significantly more likely to have histories of childhood physical or sexual abuse than those with social phobia, while the prevalence of such histories was intermediate among patients with generalized anxiety disorders; these findings held regardless of the presence or absence of comorbid anxiety or depression. Such results suggest that, consistent with other studies (
Engel et al., 1993;
Bremner et al., 1993a,
b), early abuse is a factor in the later development of anxiety disorders as well as depression.
A suggested model of vulnerability to major depressive episodes, based on both animal and clinical studies, posits that genetic factors, temperament, and trauma early and later in life markedly increase the risk of depression. When superimposed on this background of risk, stressful life events and other recent difficulties trigger a major depressive episode. Many of these effects are mediated by corticotropin-releasing factor (CRF), which plays a key role in modulating the autonomic, immune, and behavioral effects of stress; increases in CRF are associated with increased symptoms of depression and anxiety. Indeed, these findings are consistent with the repeated demonstration that mood and anxiety disorders are frequently comorbid—if not syndromally, then certainly in terms of dimensional measures of symptom severity. In fact, depression and anxiety share common effective treatments, such as SSRIs, dual serotonin-norepinephrine reuptake inhibitors, and certain forms of psychotherapy.
Is this general model consistent with preclinical findings linking early trauma with hypothalamic-pituitary-adrenal (HPA) axis functioning? In a rat model of neglect, rat pups were removed from their mothers for 3 h daily between the ages of 2–14 days, and then returned to their mothers in the animal colony for a week before weaning; this naturalistic stressor is thought to be analogous to neglect in human childhood up to the age of 4–5 years.
Pihoker et al. (1993) found that rat pups subjected to this type of maternal deprivation at the age of 10 days had significant reductions in median eminence CRF concentrations after 24 h, interpreted as an increase in hypothalamic CRF release. However, the effect was seen only during a critical time window—maternal deprivation did not reduce CRF concentrations in older, 18-day old rat pups.
The effects of early maternal deprivation appear to be remarkably persistent (
Heim et al., 2004).
Ladd et al. (1996) found that adult male rats isolated from their mothers for 6 h daily from postnatal days 2–20 (before weaning) showed increases in ACTH concentrations, both basal and induced by a mild foot shock. This effect appeared to be mediated by large increases in CRF concentrations in the median eminence of the hypothalamus, where CRF and mRNA expression were greatly increased compared to unstressed control animals. The increase in CRF mRNA expression was also seen extra-hypothalamically—for example, in the central nucleus of the amygdala and in the parabrachial region of the locus coeruleus. Moreover, direct measurements of CRF concentrations in cerebrospinal fluid (CSF) also demonstrated increases over levels in control animals.
Other intriguing changes in maternally deprived rats have also been noted. For example,
Heim et al. (2004) demonstrated that rats with histories of early maternal deprivation displayed anhedonia, as indicated by lack of reinforcement for choosing saccharin-sweetened water over plain water.
Neumaier et al. (2002) found increased 5HT
1B mRNA expression in the dorsal raphe nucleus; other pilot studies have demonstrated persistent changes including reduced hippocampal neurogenesis (unpublished observations), hippocampal mossy fiber development (
Huot et al., 2002) and GABA
A receptor binding (
Caldji et al., 2000). Gene chips are currently being used to further characterize changes in gene expression in maternally deprived animals.
What about behavioral alterations in maternally deprived rats? A hypersensitive startle to an acoustic stimulus is a well-validated marker of hyperarousal, analogous to the exaggerated startle response seen in human adults with PTSD. Plotsky et al found that maternally deprived animals showed increased acoustic startle responses compared to non-deprived and minimally separated animals (unpublished observations). Maternally deprived animals have also been shown to prefer alcohol-sucrose to water-sucrose solutions (
Huot et al., 2001), though total fluid intake was unchanged.
Interestingly, drug treatment of maternally deprived rats with paroxetine (via implantable mini-pump) has been shown to normalize—at least partially—several of the trauma-associated perturbations, both biochemical and behavioral (Plotsky et al., unpublished observations). For example, paroxetine reduced CRF mRNA expression in the paraventricular nucleus, bed nucleus of the stria terminalis, and central nucleus of the amygdala, reduced CRF concentrations in cerebrospinal fluid, normalized pituitary CRF receptor binding, attenuated the exaggerated ACTH response to startle, reversed the blunted ACTH response to exogenous CRF challenge, and decreased peripheral corticosterone levels. In contrast, paroxetine had no significant effects on these parameters in nondeprived rats. Drug treatment also normalized behaviors such as the reduced preference for sucrose and increased preference for ethanol (
Huot et al., 2001), as well as anxiety-like behavior. The time course of some of these effects is instructive: 3–4 weeks were required for the full effect of paroxetine on CRF mRNA levels in the paraventricular nucleus. However, CRF mRNA expression returned to its abnormally high level 14 days after discontination of the drug. Pilot studies with reboxetine and mirtazepine have revealed similar effects of these antidepressants (unpublished observations).
Non-human primate models of maternal deprivation have shown a similar pattern of results.
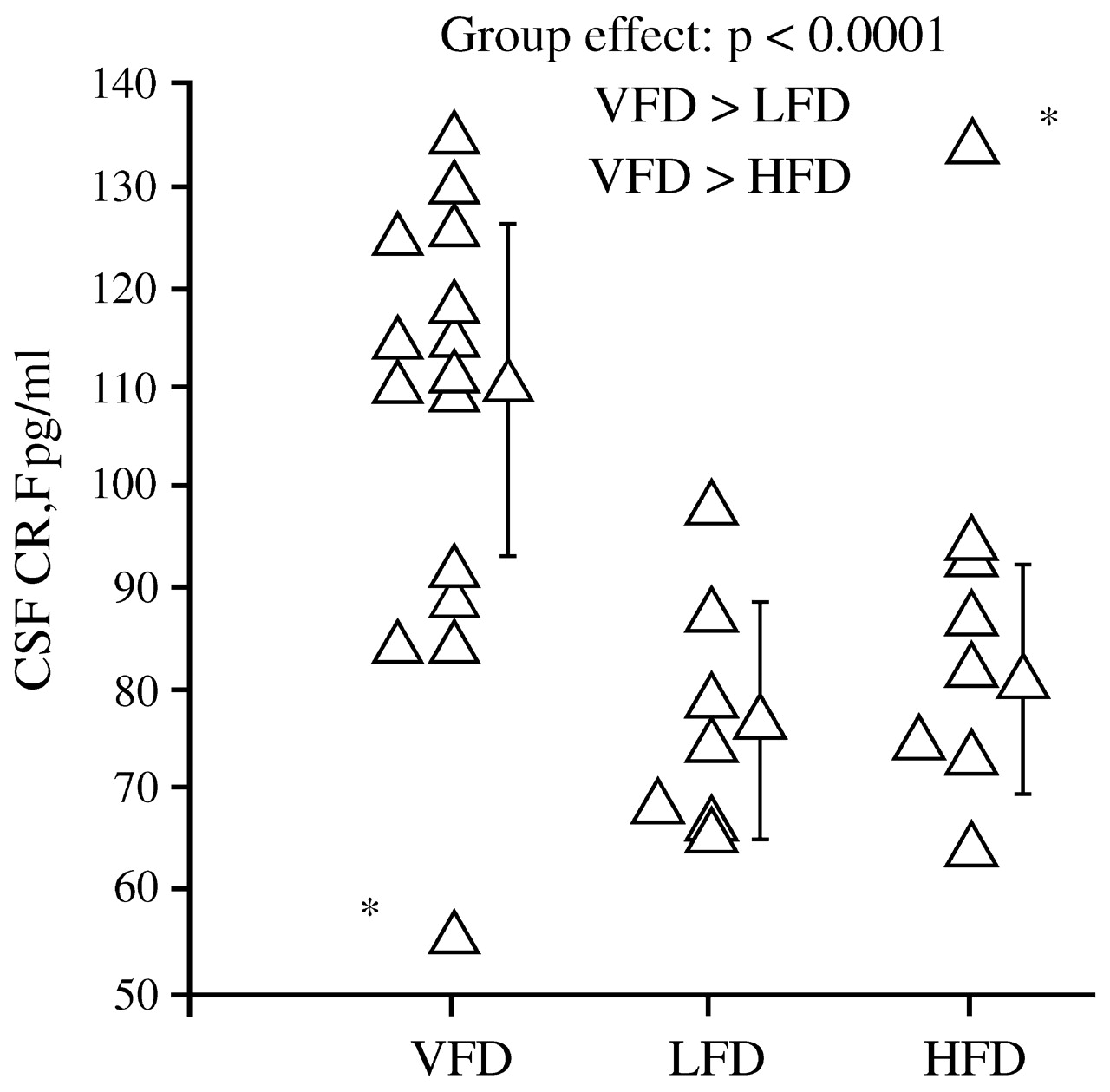
Coplan et al. (1996) found that infant bonnet macaques reared by mothers who faced variable and unpredictable foraging conditions had later persistent and significant elevations in CSF concentrations of CRF (
Fig. 6), as well as decreases in CSF concentrations of cortisol. The effects were not noted among offspring of mothers who faced high or low foraging demands, likely because these demands were predictable.
Can these preclinical findings on HPA axis responses to early life trauma be extended to the clinical arena? We believe that they can, but one must do so cautiously. In a prospective study of 49 women,
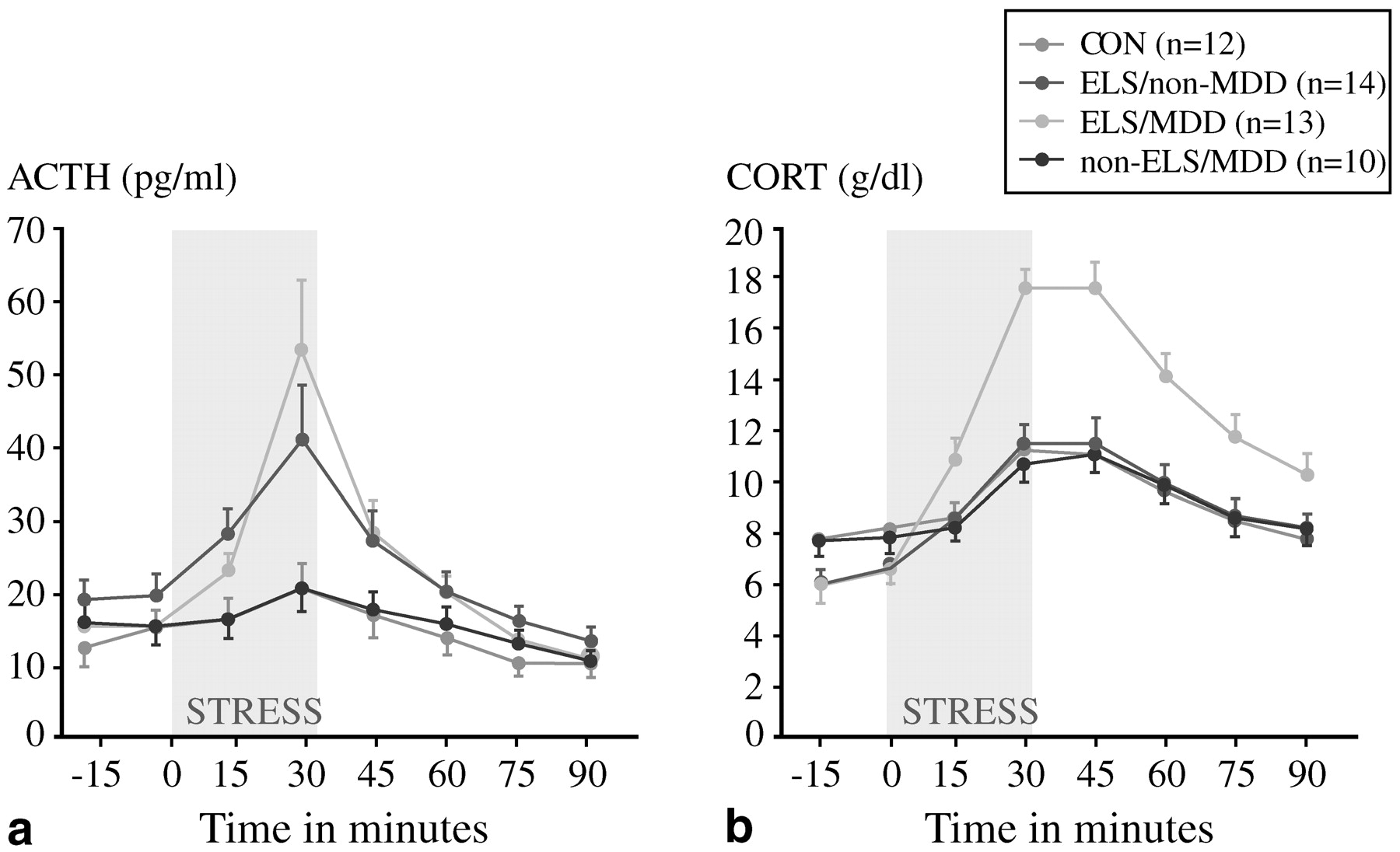
Heim et al. (2000) scrutinized four groups aged 18–45: those with no early trauma history or current psychiatric illness (controls); those with a trauma history only; those with a current episode of major depression only; and those with both major depression and early life trauma. Approximately 85% of the last group also fulfilled the diagnostic criteria for PTSD, while the rate of PTSD was 36% among those with early trauma without major depression. These women were exposed to a standard laboratory stressor, the Trier Social Stress Test, in which participants are assigned an oral presentation and mental arithmetic tasks. This stressor reproducibly raised plasma cortisol concentrations in all groups including controls, but the increases were much more dramatic among the group with both early life trauma and major depression (
Fig. 7). Whether or not they were depressed, women with early life trauma also had a much greater ACTH response to stress than controls or those with major depression only (
Fig. 7). In addition, heart rates increased modestly in controls but dramatically in subjects with early life trauma and major depression; the magnitude of the heart rate response was intermediate in the other two groups. These results are consistent with a model in which the stress system is sensitized, and subsequent stresses in adult life lead to markedly hyperactive responses. In a case-control study of 27 medication-free adults with major depression and 25 matched controls,
Carpenter et al. (2004) also found that perceived early life stress was a significant predictor of CSF CRF concentrations in both normal volunteers and depressed subjects.
Recently,
Caspi et al. (2003) reported that individuals either homozygous (s/s) or heterozygous (s/l) for the short arm of the serotonin transporter (SERT) promoter are at considerably greater risk than individuals with the l/l genotype for the development of major depression if exposed to trauma early in life. Moreover, there was a dose-response relationship for risk, such that individuals with the s/s genotype were at greater risk than those with the s/I genotype.
Several clinical studies have linked elevated CSF CRF concentrations not only with depression (
Raad-sheer et al., 1994;
Banki et al., 1987;
Nemeroff, 1989), but also with PTSD. For example,
Bremner et al. (1997a,
b) studied 11 Vietnam veterans with PTSD and found significant elevations in CSF CRF compared to controls; these elevations were seen in veterans with or without comorbid major depression.
Baker et al. (1999) subsequently measured sequentially sampled CSF cortisol concentrations and 24-h urinary free cortisol excretion in 11 combat veterans with PTSD and 12 matched control subjects, and found that CSF CRF concentrations were significantly higher among the combat veterans, though CRF levels were not correlated with PTSD symptomatology. In contrast, 24-h urinary free cortisol excretion was similar in the two groups. Interestingly, however, cortisol excretion was inversely correlated with PTSD symptoms.
Finally, what implications do these results have for treatment selection?
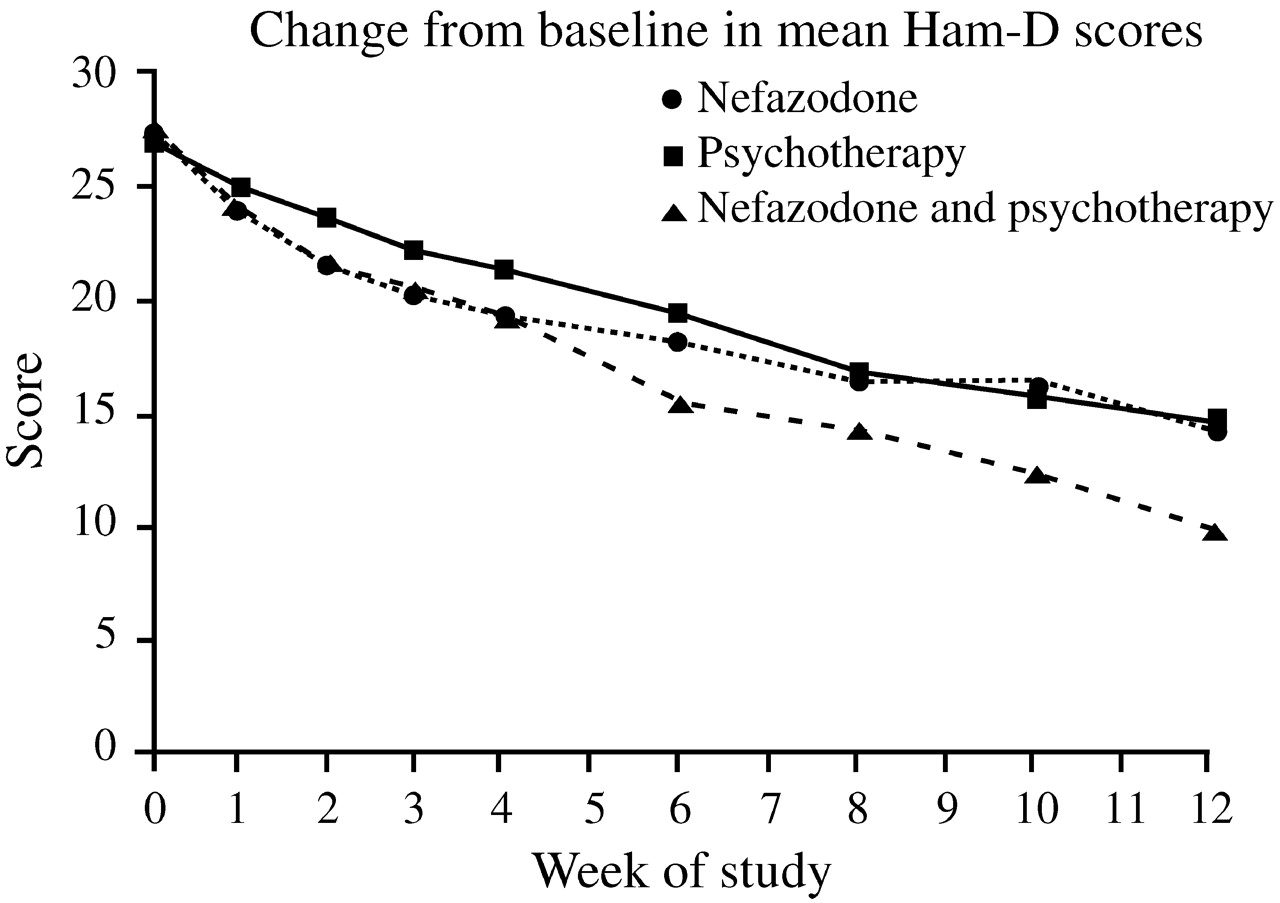
Keller et al. (2000) were among the first to compare directly the efficacy of antidepressant (nefazodone) treatment, psychotherapy, and the combination of the two in the treatment of chronic depression. This study involved 681 adults with nonpsychotic major depressive episodes at least two years long (mean duration 7.5–8.0 years) and scores ≥20 on the 24-item Hamilton Rating Scale for Depression (HAM-D). Many also had long-standing dysthymia, anxiety disorders, substance abuse disorders or personality disorders, and had undergone prior treatment with antidepressants, psychotherapy, or both. These patients were randomized to receive either the cognitive-behavioral-analysis system of psychotherapy (CBASP) (
McCullough, 1984), delivered in 16–20 sessions over 12 weeks, or nefazodone (mean dose 460–466 mg daily), or both treatments. After 12 weeks, remission rates (HAM-D ≤8) among study completers were 22% in the nefazo-done group, 24% in the CBASP group, and 42% in the combined group (
p < 0.001 vs either single modality) (
Fig. 8). These findings were consistent with the wide-spread clinical impression that combination treatment is superior to single modalities.
A secondary analysis (
Nemeroff et al., 2003) of the data set from the foregoing study revealed several novel findings. First, there is an extremely high prevalence of early life trauma in this chronically depressed population. Only about one-third of the patients had no trauma history. Overall, 32–35% had suffered parental loss before age 15 years, 40–48% physical abuse, 15–19% sexual abuse and 7–12% neglect. Patients with histories of trauma had dramatically poorer responses to antidepressant treatment than to psychotherapy or combined treatment. The differences in remission rates were even more striking—32% on antidepressant therapy, 48% on psychotherapy, and approximately 52% on combined therapy. Given that treatment of maternally deprived animals with SSRIs can reverse many of the adverse biochemical and behavioral effects of trauma, it would be desirable to conduct a similar study with an SSRI in humans; a trial involving fluoxetine is currently underway.
5. WHAT CAN IMAGING STUDIES OF DEPRESSION TEACH US ABOUT FAILED ADAPTATION TO CHRONIC EMOTIONAL STRESS AND ANXIETY DISORDERS?
Research on functional brain imaging in depression (and on the biology of depression in general) has “matured” much more than similar research in PTSD. Not only can we learn a great deal from the depression literature that is relevant to PTSD, but it is now well established that these syndromes are frequently comorbid, and that certain antidepressants, the SSRIs in particular, are effective for both conditions.
Depression may be conceptualized as an end-product of failed adaptation to chronic emotional stress. The individual, who begins with a given set of biological risks (e.g., female sex, certain gene polymorphisms, temperament, and reactivity of the HPA axis) is subjected to a variety of exogenous stressors, which act to destabilize the usual homeostatic state of the brain circuits that are involved in mood regulation. The result is a depressive episode, and the goal of treatment is to restabilize the key brain circuits. Defining these brain circuits and their perturbations as precisely as possible by separating their putative components experimentally is a major research challenge.
Do depressed patients differ from healthy individuals in their acute adaptive responses to stress? Several studies have used provoked sadness and neuroimaging studies to determine changes in regional brain activity in response to this sad mood challenge. Normal individuals, depressed patients, and those thought to be at risk for depression may be studied in this way.
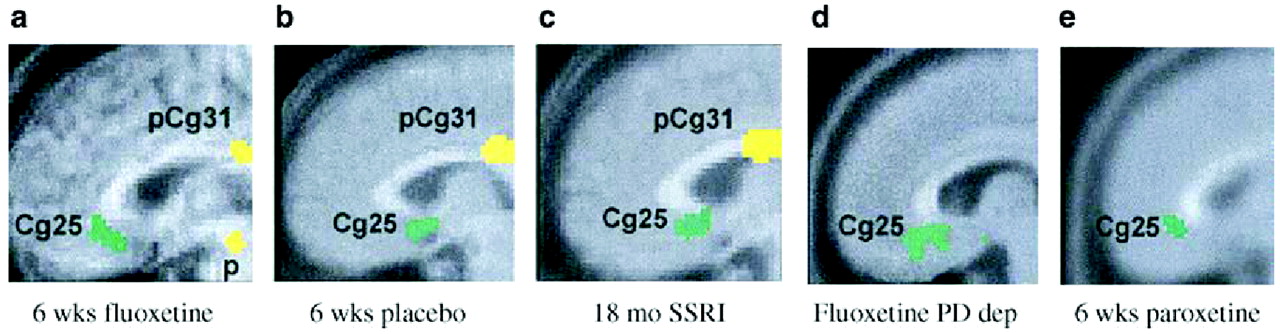
Mayberg et al. (1999) asked eight healthy right-handed euthymic women to prepare scripts of sad personal experiences and then to allow themselves to feel sadness while undergoing [
15O]H
2O positron emission tomography (PET). Limbic-paralimbic blood flow was increased, particularly in the subgenual cingulate (Brodmann's area 25) and the anterior insula, while neocortical blood flow (in the right dorsolateral prefrontal and inferior parietal areas) was reciprocally decreased. This pattern was consistent with the well-known clinical observation that intense sadness is often accompanied by cognitive difficulties.
A very different pattern was seen in depressed patients.
Liotti et al. (2002a,
b) compared regional blood flow changes after the same sad mood challenge among acutely depressed patients (mean HAM-D score 21.3), euthymic patients who were in remission (mean HAM-D score 4.8 without medications), and normal volunteers. Decreases in blood flow in the medial orbitofrontal cortex (area 10/11) and a variety of other areas were virtually identical in both the remitted and symptomatic depressed patients, and differed substantially from control subjects. Remitted patients (but not acutely depressed patients or controls) exhibited decreased blood flow in the pregenual anterior cingulate (area 24a). Conversely, the depressed groups lacked the increase in area 25 and the decrease in right prefrontal cortex that were observed in the controls. More recently,
Kruger et al. (2003) studied bipolar patients and found patterns of cerebral blood flow in response to sad mood challenge that were similar whether the patients were acutely depressed or in remission. Taken together, these results suggest that the unique changes seen across patient subtypes, irrespective of clinical state, may be trait markers of a depressive diathesis.
Can depressive illness also be conceived of as the product of more chronic maladaptive changes to emotional triggers? Most depressed participants in neuroimaging studies are recruited well after the onset of symptoms, and may present at various points in the course of the brain's attempts at adaptation. Moreover, patients recruited from tertiary care centers are likely to be different from those recruited by newspaper advertising, and their clinical states can range from complete failure of adaptation to partial adaptation or hypersensitive adaptations. The heterogeneity of neuroimaging results reported in the literature is therefore not surprising. In fact, this baseline variability may be a potential clue to understanding differences in both the clinical and brain response patterns across different treatment modalities (
Mayberg, 2003).
In a placebo-controlled, double-blind trial,
Mayberg et al. (2000a,
b) explored changes in brain glucose metabolism using PET in unipolar depressed male inpatients after 1 week and 6 weeks of treatment with fluoxetine. Patients receiving active fluoxetine had a similar pattern of changes in glucose metabolism after 1 week of treatment: increases in the hippocampus and brainstem and decreases in the posterior cingulate, striatum, and thalamus. By 6 weeks, however, fluoxetine responders could be distinguished from non-responders by decreases in limbic and striatal glucose metabolism (subgenual cingulate, hippocampus, insula, and pallidum) and reciprocal increases in brainstem and dorsal cortical areas (prefrontal, parietal, anterior, and posterior cingulate). In contrast, the patterns seen during the first week persisted in the non-responders. Interestingly, regional changes were not merely corrections of pretreatment abnormalities. Some regions showed changes in the direction of reversing baseline abnormalities, while others were new effects in regions showing apparently normal pretreatment activity. These findings suggest that successful treatment is not a simple matter of absolute changes, but instead entails a more complex adaptation of multiple brain regions to a new setpoint. Conversely, clinical treatment failure may be the result of an inability to institute or maintain these adaptations. The metabolic changes with response were not specific to fluoxetine treatment, because they were also noted among the placebo responders in the study. The time course and localization of these changes were also consistent with the data obtained by several groups on upregulation of the cyclic AMP second messenger system and increased expression of brain-derived neurotrophic factor (BDNF) in association with antidepressant treatment (
Vaidya et al., 1997;
Duman et al., 1999;
Chen et al., 2001). Finally, the changes appear to persist over time; decreased activity in area 25 was seen 18 months later (
Mayberg, 2003;
Liotti et al., 2000) (
Fig. 9—
Mayberg et al., 2000a,
b;
Mayberg et al., 2002;
Liotti et al., 2002a,
b;
Stefurak and Mayberg, 2003;
Goldapple et al., 2004a,
b).
Kennedy et al. (2001) carried out a similar study of brain glucose metabolism among 13 depressed male patients before and after 6 weeks of therapy with paroxetine 20–40 mg daily, comparing the patterns obtained with those of 24 healthy male volunteers. All patients responded to treatment (HAM-D scores reduced by at least 50%). With successful treatment, glucose metabolism increased in the prefrontal cortex, parietal cortex, and dorsal anterior cingulate, and decreased in left anterior and posterior insula (though it remained higher than in controls) and right hippocampus and parahippocampal regions. Anterior cingulate metabolism, already hyperactive before treatment began, increased even further after treatment. Reanalysis of the data obtained by Kennedy et al showed that the subgenual cingulate (area 25) was also suppressed with paroxetine treatment (
Fig. 9).
Changes in subgenual cingulate may be central to recovery from depression due to its very high concentrations of serotonin transporters, a marker of presynaptic serotonin-containing nerve terminal density, and a primary target of SSRIs. This region also has reciprocal cortical and subcortical connections to the hippocampus, hypothalamus, amygdale, brainstem, and frontal cortex—regions involved in regulating circadian and autonomic as well as cognitive functions disturbed during a major depressive episode (
Freedman et al., 2000). Modulation of this area therefore has the potential for broad cortical effects (
Ongur et al., 1998;
Barbas et al., 2003).
It might be tempting to conclude that successful treatment of depression occurs through a prerequisite final pathway—cortical normalization and limbic suppression. This is apparently not the case.
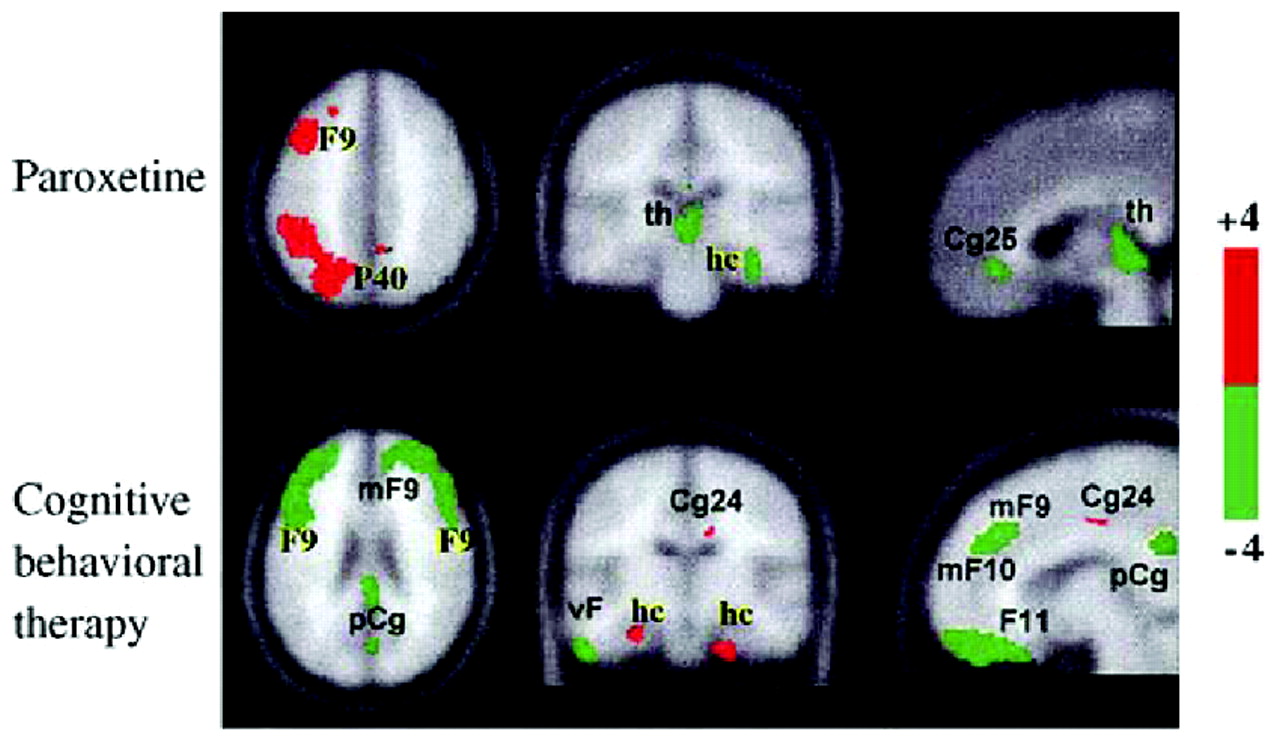
Goldapple et al. (2004a,
b) recruited 17 depressed patients with no comorbid psychiatric illnesses and studied changes in cerebral metabolism before and after they received 15–20 sessions of cognitive-behavioral therapy (CBT). Fourteen patients completed the study and all had significant clinical improvement (mean posttreatment HAM-D score 6.7). The metabolic changes among these patients were in many regions precisely the opposite of those seen with paroxetine treatment: increases in the hippocampus and dorsal cingulate and decreases in the dorsal, ventral, and medial frontal cortex. Dorsal cingulate and medial frontal regions were unique targets of CBT. Similarly, the subgenual cortex, thalamus, and brainstem were targeted with medication but were unaffected by the CBT (
Fig. 10). The decreases in cortical metabolism are consistent with the idea that CBT has the effect of decreasing rumination about emotionally salient aversive events and memories.
These results imply that instead of going through a single final common pathway, different treatment modalities modulate the system in different ways—drug treatment primarily via a subcortical approach (“bottom-up”) and cognitive approaches primarily via a cortical approach (“top-down”;
Fig. 11). They also suggest that subtyping depression according to the brain's baseline metabolic state might be an effective method of selecting the best treatment modality for a given individual.
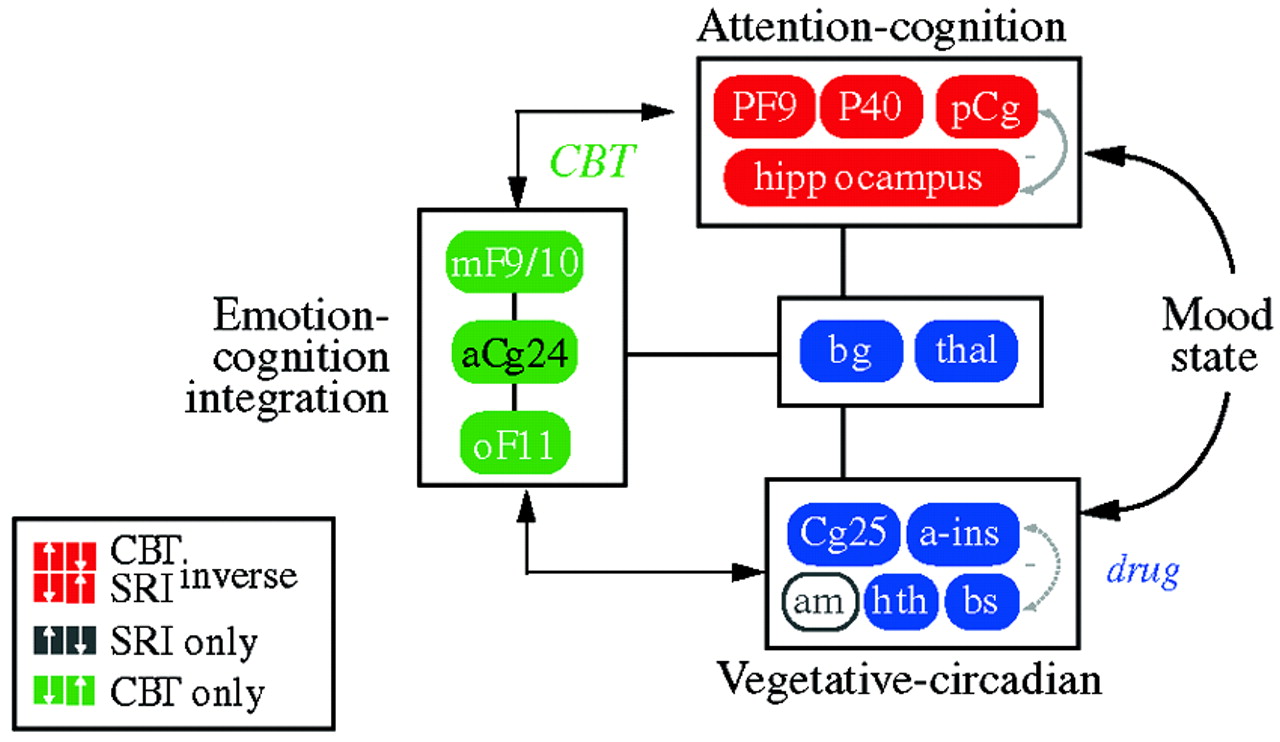
In an attempt to define common denominators of successful treatments,
Seminowicz et al. (2004) developed a simplified 7-region model derived from the results of these treatment studies; this was mathematically tested using structural equation modeling. The simplified model examining regional interactions of medial frontal (MF10), orbital frontal (OF11), lateral prefrontal (PF9), subgenual cingulate (Cg25), anterior cingulate (Cg24), hippocampus (hc), and thalamus (thal) activity prior to treatment succeeded in accommodating all patient cohorts tested—a total of 119 patients from two institutions. Patients who went on to respond to CBT showed a mainly cortical pattern of aberrant brain connections at baseline, while drug responders showed a combined limbic-cortical pattern. Patients who were eventual nonresponders to medication showed a third distinct baseline network pattern suggestive of a more complex limbic-subcortical disturbance. The modeling results lay the groundwork for future clinical trials examining treatment response based on brain subtypes.
Finally, can the interactions between Brodmann's area (BA) 25 and frontal cortex be useful in detecting populations who are vulnerable to depression?
Keightley et al. (2003) examined activation patterns in response to sad mood challenge among healthy college students with no personal or family history of depression who were selected for resilience (low scores on the dispositional Depression facet of Neuroticism [N3] and high scores on the Positive Emotions face of Extraversion [E6] on the NEO Personality Inventory-Revised) or for the reverse pattern—high N3 scores and low E6 scores. In response to the mood challenge, all subjects had decreases in lateral frontal cortical activation and increases in the activation of BA 25, but the high-N3 subjects had low medial frontal activation that resembled the pattern seen in depressed patients. These data suggest that this paradigm is highly sensitive to risk of depression, a hypothesis that requires testing in other vulnerable subject groups.
The foregoing results illustrate the value of a meticulous dissection of the different neural circuits involved in psychiatric illness and its treatment. While this approach may initially appear to be oversimplified, it has the advantage of allowing the complex mechanisms that underlie vulnerability, treatment, and relapse to be clarified.
6. NEURAL CIRCUITS, MEMORY, AND PTSD
The neural circuitry implicated in PTSD probably involves complex interactions between the thalamus (a gateway for sensory inputs), the hippocampus (which is involved in short-term memory and probably fear of the context of an event), the amygdala (which is involved in conditioned fear responses), the posterior cingulate, parietal and motor cortex (which are involved in visuospatial processing and assessment of threat), and the medial prefrontal cortex, including the anterior cingulate, orbitofrontal, and subcallosal gyrus (which is believed to extinguish more primitive subcortical responses).
The effects of stress on the hippocampus have been recognized since the 1990s, but the effects of stress on neurogenesis are of more recent vintage:
Woolley et al. (1990) demonstrated that exposure of adult rats to excess corticosterone (equivalent to cortisol in humans) reduced dendritic branching in specific neuronal populations of the hippocampus, suggesting early stages of degeneration, and
Gould et al. (1998) found decreased neurogenesis in socially subordinated non-human primates compared with dominant animals. Moreover,
Duman et al. (2001) demonstrated that chronic administration of electroconvulsive therapy or a variety of antidepressants, but not other psychotropic drugs, to adult rodents upregulated hippocampal neurogenesis,
Santarelli et al. (2003) found that irradiation of the hippocampus in mice prevented the neurogenic and behavioral effects of fluoxetine or imipramine, suggesting that hippocampal neurogenesis may be one of the mechanisms of action of antidepressants in the treatment of stress-related disorders.
Bremner et al. (1993a,
b) used neuropsychological testing (paragraph recall) as a probe of hippocampal function in 26 Vietnam veterans with combat-related PTSD and 15 matched healthy control subjects. Compared to controls, the combat veterans had decreased immediate and delayed recall as well as percent retention, but IQ scores were similar in the two groups.
Bremner et al. (1995a,
b) also found similar results among adult survivors of severe childhood physical or sexual abuse who presented for psychiatric treatment.
The results of these studies introduced the possibility that stress in humans may also lead to hippocampal damage (
Bremner, 1998;
Pitman, 2001;
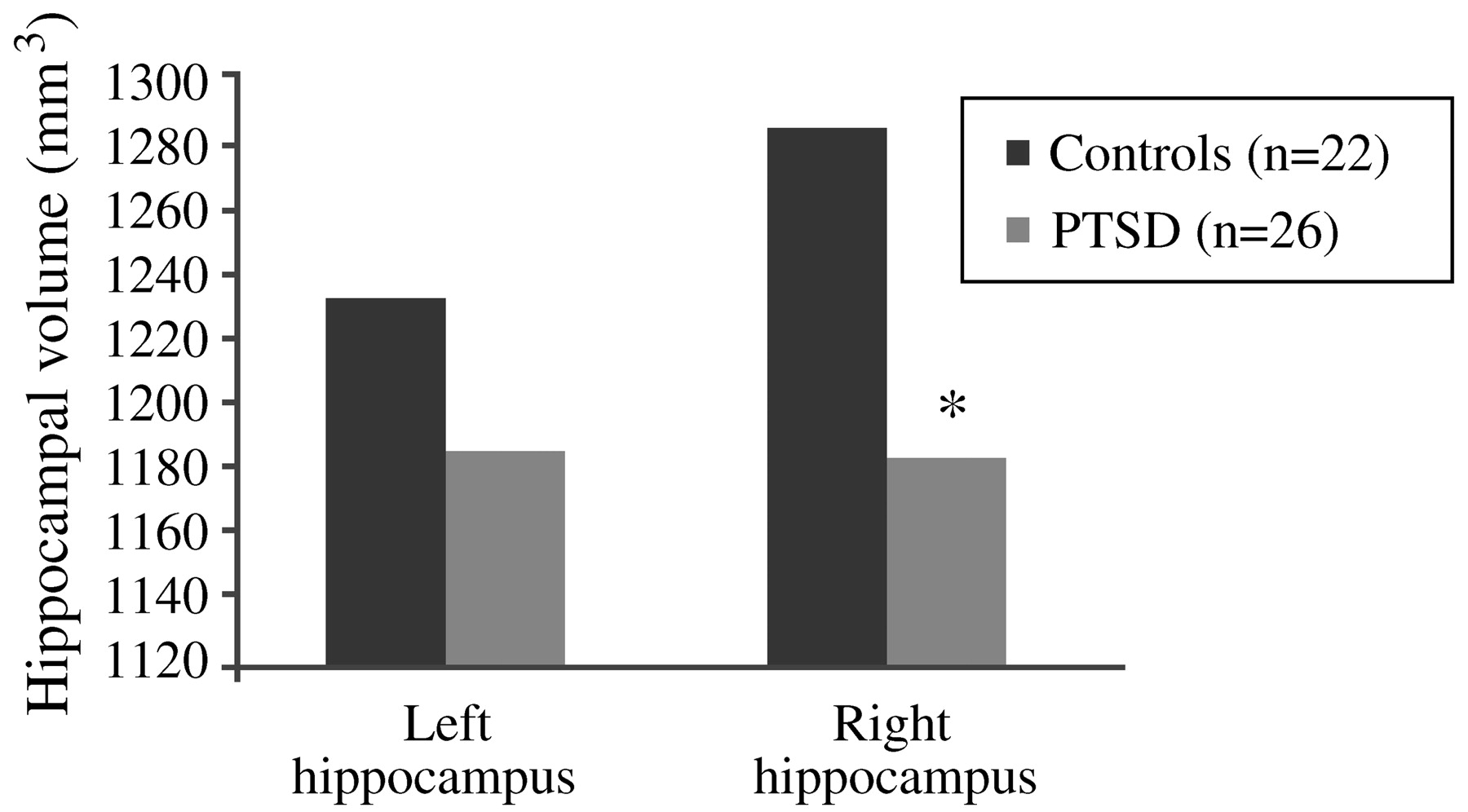
Bremner, 2002). The first neuroimaging study in PTSD was carried out using magnetic resonance imaging (MRI) to measure hippocampal volumes in 26 Vietnam combat veterans with PTSD and 22 matched control subjects. Mean right hippocampal volume was 8% smaller in the veterans than in the control subjects (1,184 mm
3 vs 1,286 mm
3;
p < 0.05;
Fig. 12), and was correlated with short-term memory deficits on the Wechsler Memory Scale (r = 0.64;
p < 0.05). Decreases in right hippocampal volume in the PTSD patients were associated with deficits in short-term memory. Left hippocampal volume was also decreased in the veterans, but the difference did not reach statistical significance (
Bremner et al., 1995a,
b). Findings of smaller hippocampal volume and/or a reduction in
N-acetylaspartate (NAA) in the hippocampus—a marker of neuronal integrity—in adults with chronic, long-standing PTSD have been replicated several times in the published literature (
Gurvits et al., 1996;
Bremner et al., 1997a,
b;
Stein et al., 1997a,
b;
Freeman et al., 1998;
Schuff et al., 2001;
Bremner et al., 2002;
Villareale et al., 2002).
Using [
15O]H
2O PET,
Bremner et al. (1999a,
b) measured cerebral blood flow in 22 women with histories of childhood sexual abuse while they listened to scripts that were either neutral or traumatic (personalized accounts of childhood sexual abuse events). Compared with those without PTSD, the traumatic scripts evoked increased blood flow in the posterior cingulate, anterior prefrontal cortex, and motor cortex, but decreased blood flow in the right hippocampus, visual association cortex, and the subcallosal gyrus (area 25) and adjacent anterior cingulate (area 32) of the medial prefrontal cortex. In tandem with this, women with PTSD (but not those without the diagnosis) had increased PTSD symptom scores as they listened to the traumatic scripts. This pattern of activation was similar to that observed in Vietnam veterans who were exposed to slides and sounds evoking combat situations (
Bremner et al., 1999a,
b). These results are consistent with a general model of cortical failure to suppress exaggerated subcortical reactions to stress.
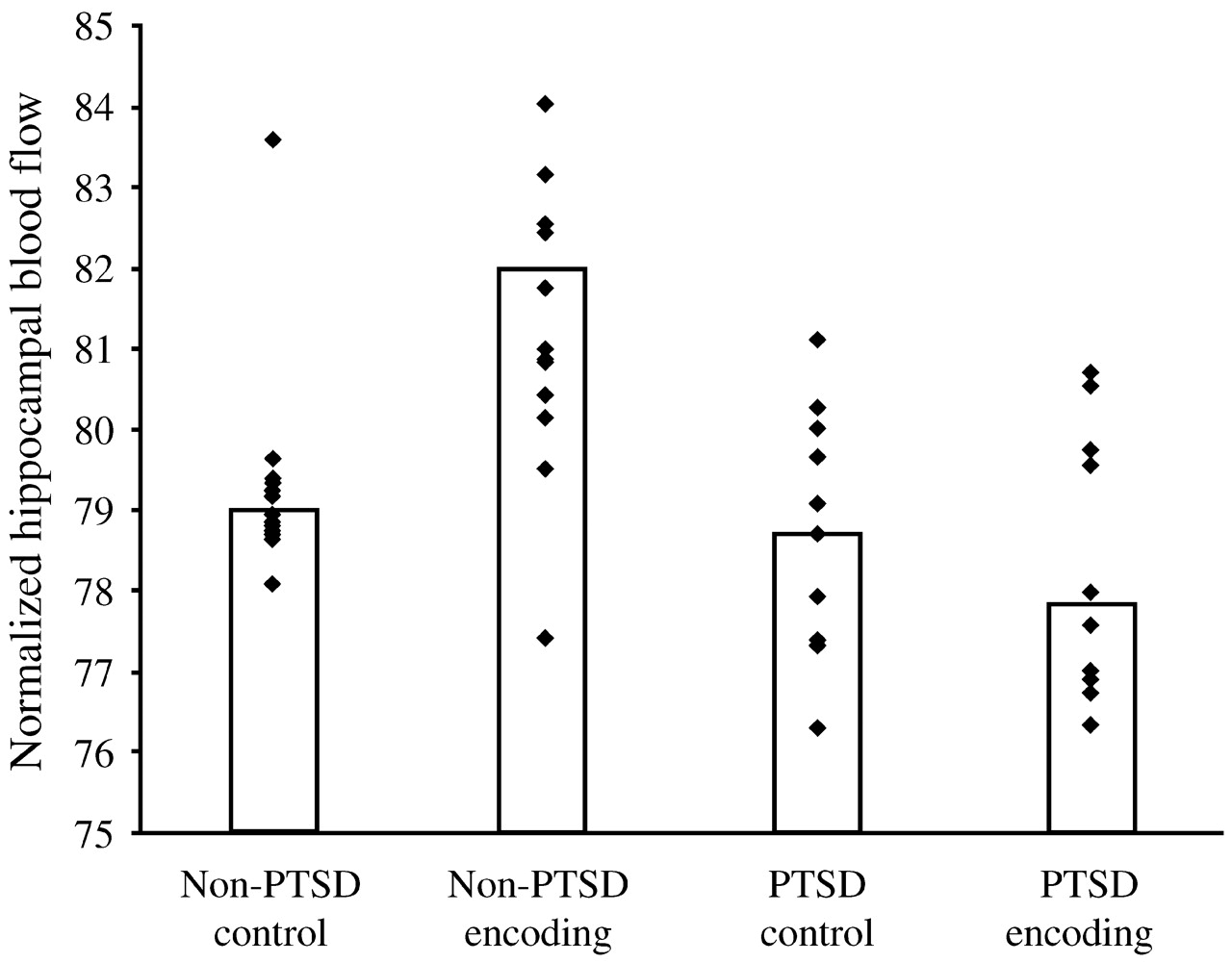
Are the hippocampal changes seen in PTSD actually a function of the early childhood traumas seen in so many PTSD patients, or are they specific to PTSD itself? In a more recent study,
Bremner et al. (2003) studied 33 women with early childhood sexual abuse and PTSD (
n = 10), early childhood sexual abuse without PTSD (
n = 12) and no abuse or PTSD (
n = 11). Hippocampal volumes (as measured with MRI) among the women with abuse histories and PTSD were 16% smaller than among those with abuse histories without PTSD, and 19% smaller than among the normal controls, with the right hippocampal volume somewhat more diminished than the left. The women were also given two tasks: to count the number of times they heard the letter “d” in a paragraph that was read aloud to them (control condition) and to recall as much as possible of a different paragraph they had been read (verbal memory encoding task). In contrast to the women with abuse histories without PTSD, the women with PTSD did not show increased blood flow to the hippocampus during the verbal encoding memory task. Thus, there was a failure of left hippocampal activation with a memory task among women with abuse-related PTSD, which remained significant after controlling for differences in hippocampal volume (measured with MRI in the same subjects) (
Fig. 13).
Shin et al. (2004) have also demonstrated a failure of hippocampal activation with a declarative memory task in PTSD.
However, studies in children with PTSD (
De Bellis et al., 1999;
Carrion et al., 2001;
De Bellis et al., 2001) and in adult subjects with new-onset PTSD (
Bonne et al., 2001;
Notestine et al., 2002) have not demonstrated a smaller hippocampal volume, suggesting that chronic PTSD is required for the effect. One study of monozygotic twins discordant for exposure to trauma found a correlation between PTSD symptoms and hippocampal volume in the unexposed twin, suggesting a genetic contribution to smaller hippocampal volume (
Gilbertson et al., 2002). However, another unpublished twin study of twins discordant for PTSD has shown a pattern of hippocampal volume that is consistent with a combined genetic and environmental effect.
Preclinical studies have suggested that stress-induced lesions of the hippocampus attenuate its normal inhibitory effect on the HPA axis. The cascade of consequences includes enhanced release of CRF from the hypothalamus, a blunted ACTH response to CRF challenge, increased peripheral plasma cortisol concentrations, and—similar to what is observed in depression—resistance to the usual suppressive effects of dexamethasone. However, there are puzzling inconsistencies in findings among patients with PTSD; for example, some studies—but not all—have found decreased urinary cortisol levels (
Mason et al., 1986;
Yehuda et al., 1995) or “super” suppression of cortisol by low doses of dexamethasone (
Yehuda et al., 1993;
Stein et al., 1997a,
b).
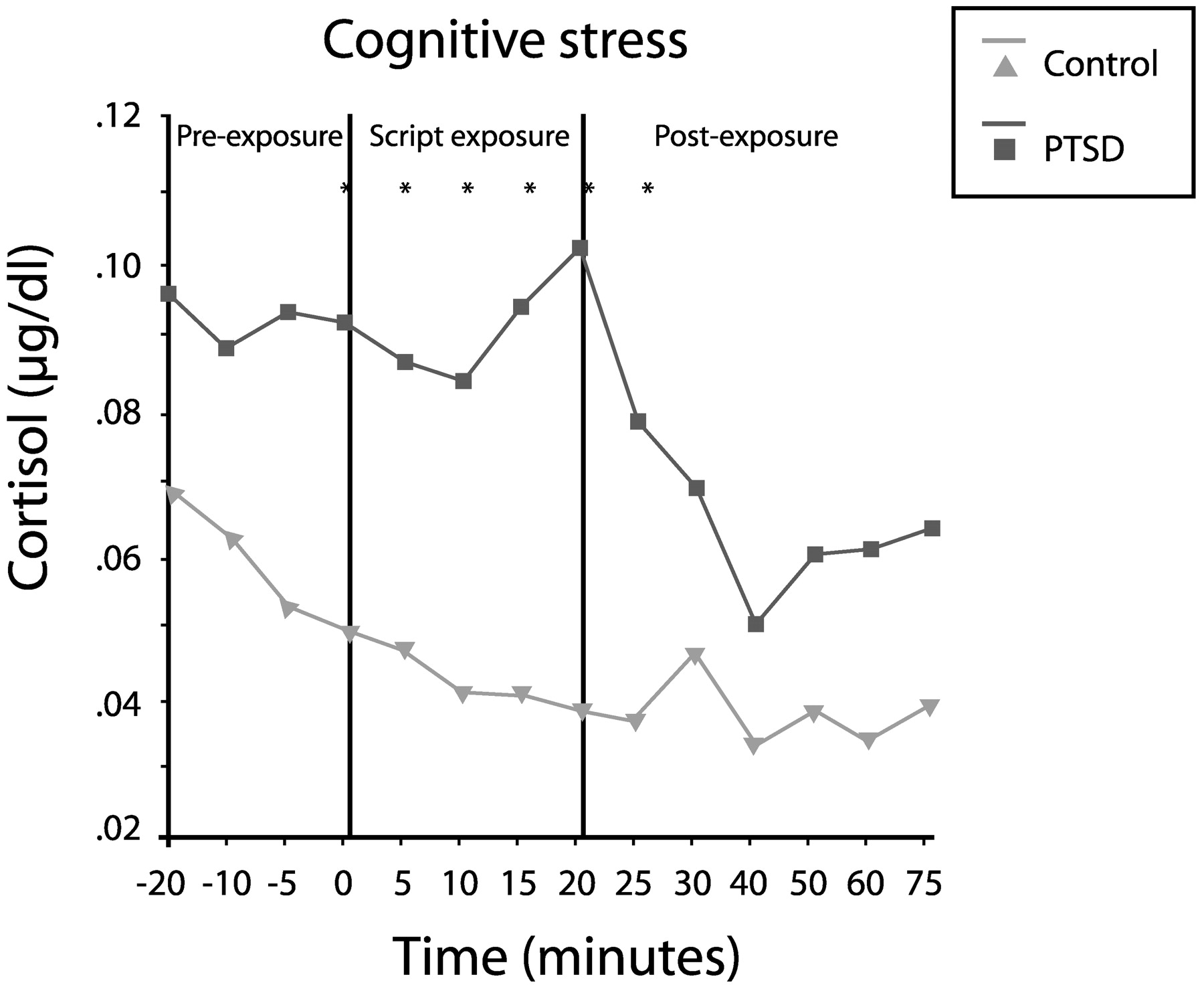
Elzinga et al. (2003) measured cortisol responses to traumatic reminders (via personalized trauma script) in 24 women with abuse histories in the presence or absence of PTSD. Cortisol concentrations in the patients with PTSD were 60% higher during anticipation of the script reading (from 20 min prior to the reading), 122% higher during the reading and 69% higher during the recovery phase (up to 75 min later;
Fig. 14). Thus, PTSD appears to involve low baseline cortisol levels, but an exaggerated release of cortisol in response to a stressor.
Paroxetine has been shown to be highly effective in the treatment of PTSD (
Marshall et al., 2001;
Marshall et al., 1998;
Tucker et al., 2001).
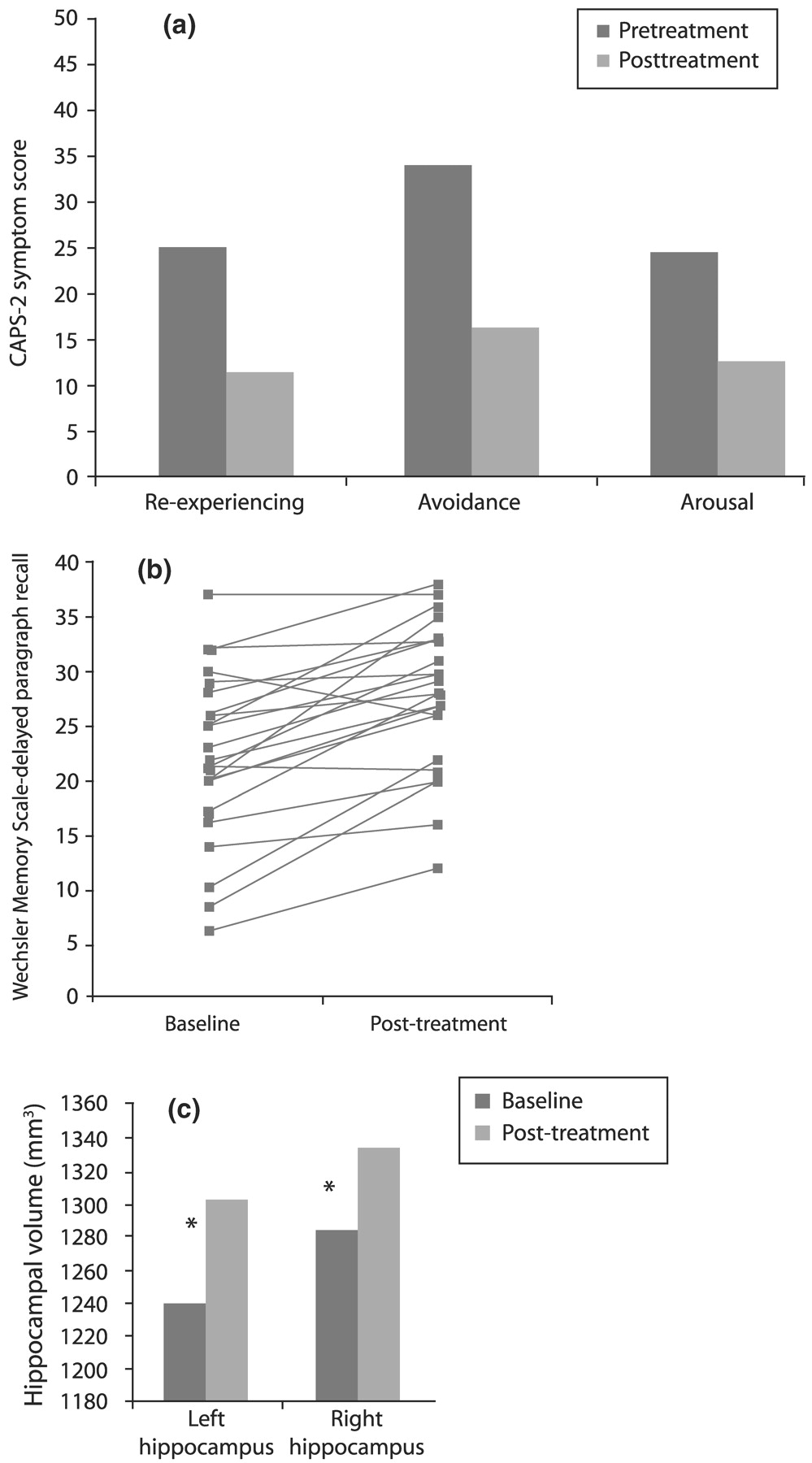
Vermetten et al. (2003) measured hippocampal volume and verbal declarative memory (a hippocampus-based task) in 28 patients with PTSD before and after 9–12 months of paroxetine treatment. Twenty-three patients completed the study, and 20 had MRI for assessment of hippocampal volume. Paroxetine significantly reduced symptoms in all three domains of the CAPS-2 (
Fig. 15(a)), improved verbal declarative memory (
Fig. 15(b)) and increased left and right hippocampal volume by 5.6% and 3.7%, respectively (
p < 0.05;
Fig. 15(c)). The effects of other medications on these parameters in PTSD remain to be explored.
7. COGNITIVE BEHAVIORAL APPROACHES TO THE TREATMENT OF PTSD: WHAT WORKS?
Like re-experiencing, hyperarousal and avoidance symptoms, negative cognitions are ubiquitous immediately after an individual suffers a trauma (
Foa and Jaycox, 1999). Usually, however, subsequent everyday life experiences gradually correct these negative cognitions, and the traumatized individual has the opportunity to regain a sense of competence and safety in the world. In contrast, those who make extensive use of avoidance and numbing will also avoid the very experiences that could have corrected their cognitive distortions. Thus, these individuals may be at higher risk for the development of PTSD.
Foa and Jaycox (1999) have suggested that two particular groups of erroneous cognitions are associated with the later development of PTSD: that the world is extremely dangerous (for example, that no place is safe and that people are untrustworthy) and that the sufferer is extremely incompetent (e.g., that others would have been able to prevent the trauma somehow, and that the sufferer's PTSD symptoms are a clear sign of weakness).
Foa et al. (1999a,
b) explored self-blame and negative thoughts about the self and the world among three groups of subjects with PTSD, trauma exposure without PTSD, or no trauma exposure. The group with chronic PTSD had significantly higher levels of these negative cognitions than the other two groups. While this study was not longitudinal and therefore could not address causality, the similarity between the non-traumatized subjects and those who had been traumatized but did not develop PTSD suggests that the negative cognitions may be more important than the experience of trauma per se in driving the development of PTSD.
Although most therapists working with traumatized individuals use psychodynamic or supportive counseling approaches—for which there are no efficacy data—most studies of PTSD treatment outcomes have explored cognitive-behavioral therapies, which fall into three general subtypes. In exposure therapies such as systematic desensitization and flooding, patients confront their fears, object, situation, memories, and images without being as overwhelmed as they had anticipated. These experiences of exposure thus serve to disconfirm and correct cognitive distortions harbored by the patient. The anxiety management component includes a variety of techniques such as relaxation, controlled breathing, and self-distraction (thought stopping); patients carry out exercises designed to improve their anxiety management skills. Finally, cognitive therapy identifies and challenges dysfunctional and erroneous cognitions, aiming to replace them with more functional and realistic cognitions. This procedure can be said to contain elements of exposure in the sense that the patient is exposed to the traumatic memories and thoughts and must process them differently in order to recover.
The clinical practice guidelines developed by the International Society for Traumatic Stress Studies (
Foa et al., 2000) suggest that exposure therapy is the most empirically supported for PTSD, but cognitive therapy (e.g.,
Resick et al., 2002) and interpersonal psychotherapy (
Bleiberg and Markowitz, 2005) have also been shown to be effective for this disorder.
Keane et al. (1989) conducted the first study of implosion therapy (flooding) combined with relaxation in Vietnam veterans, a difficult population, and obtained a modest improvement in PTSD symptoms compared to wait-listed control subjects.
Foa and Rothbaum (1998) developed a prolonged exposure (PE) therapy program consisting primarily of prolonged, repeated exposure to the traumatic memory and repeated in vivo exposure to situations that are being avoided because of trauma-related fear; the treatment also contains elements of breathing retraining and psychoeducation about common reactions to trauma. (For a comparison between the efficacy of imaginal and in vivo exposure, see
Bryant et al., 2003.)
Stress inoculation training (SIT) (
Meichenbaum, 1974) contains elements of relaxation training, thought stopping, self-guided dialogue, cognitive restructuring and covert modeling, and role play.
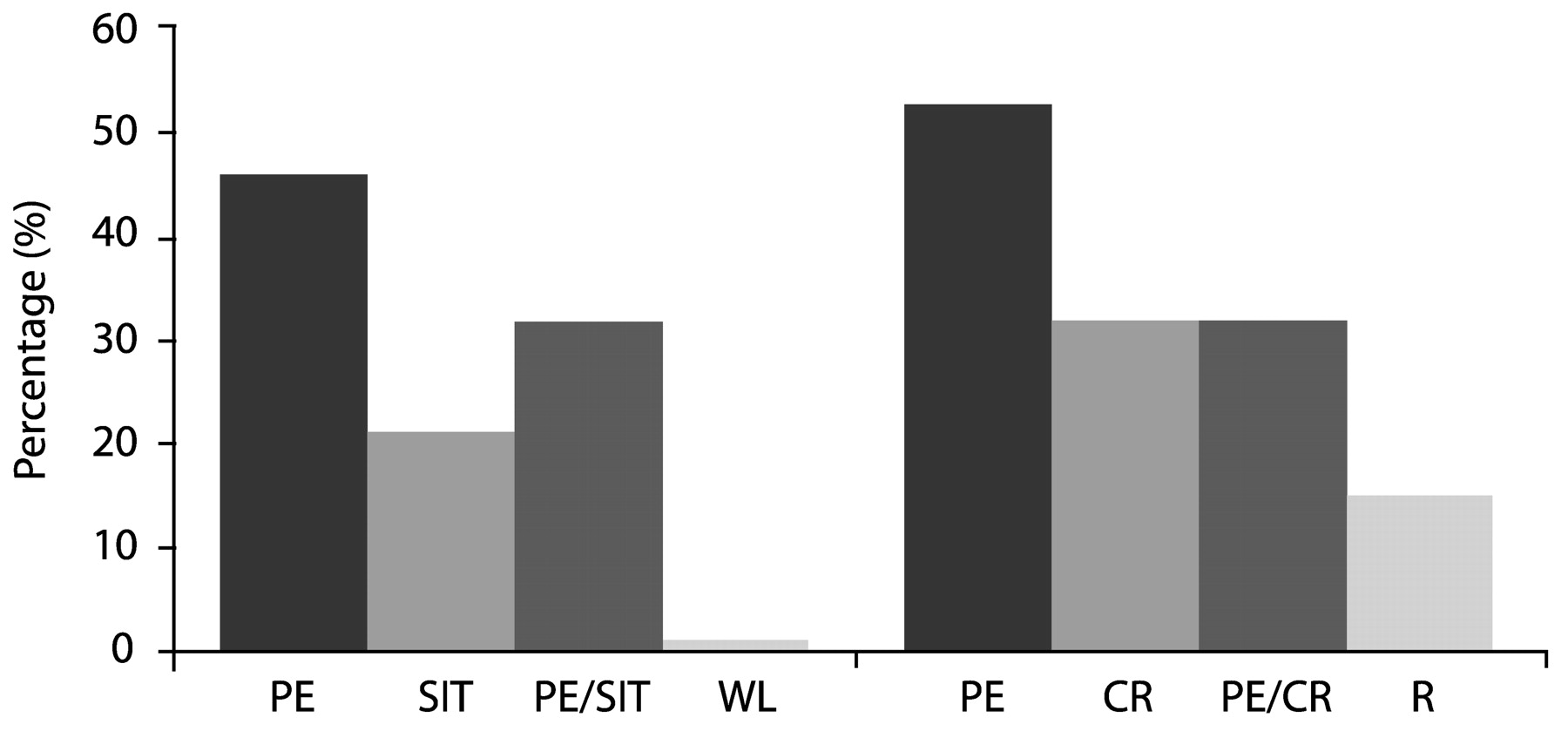
Foa et al. (1991) compared SIT, PE, supportive counseling and waiting list conditions in 45 rape victims with PSTD, and found that while PTSD symptoms improved in all groups, SIT produced significantly more improvement immediately after treatment (55% symptom reduction, vs 40% for PE, 26% for supportive counseling, and 20% for waiting list controls). However, PE was superior at 3 months post-treatment—PTSD symptom reduction at this point was 60% for this modality compared with 49% for SIT and 36% for supportive counseling.
In a second study,
Foa et al. (1999a,
b) compared the efficacy of PE, SIT, both treatments combined, and waiting list among 96 female assault victims with chronic PTSD. These patients, whose post-assault intervals averaged 5 years, had a high prevalence of comorbid depression, but any medication regimens were to remain unchanged throughout the course of the study. Treatment consisted of nine sessions over 5 weeks. All three active treatments significantly reduced both PTSD and depressive symptoms to a similar extent; contrary to initial expectations, combined therapy did not improve outcomes over either single modality. In fact, exposure therapy alone yielded an effect size of 1.92, larger than SIT (1.61) or the combination (1.50). The gains were maintained through a 12-month followup period.
Using a slightly different design in which nine sessions of exposure therapy were extended over 9 weeks, optionally augmented by three additional sessions for those patients with a partial treatment response (<70% improvement), Foa et al. (in press) obtained similar results: adding cognitive restructuring to exposure therapy did not produce a significant benefit over that conferred by exposure therapy alone. One possible reason for this result is that exposure therapy alone is successful in modifying negative cognitions.
Similar results in favor of exposure therapy were found by
Marks et al. (1998) among 87 male and female subjects who had suffered a variety of traumas. These authors compared the efficacy of 10 sessions of prolonged imaginal and live exposure, cognitive restructuring alone, the combination of the two, or relaxation therapy without prolonged exposure or cognitive restructuring. Both PE and cognitive restructuring were effective, but the combination did not offer increased benefits. In fact, PE produced over 50% improvement in end-state functioning, more than any of the other conditions including combination treatment (
Fig. 16).
Eye movement desensitization and reprocessing (EMDR) (
Shapiro, 1995) is a therapy which involves accessing traumatic images and memories, evaluating their aversive qualities, and generating and focusing on alternative cognitive appraisals of these images and memories while performing sets of saccadic eye movements. Comparing EMDR with exposure therapy in female rape victims with PTSD,
Rothbaum and Astin (2002) found that the latter produced a numerically but not statistically higher treatment response rate at 6 months follow-up (defined as a CAPS score reduction of ≥50%, a Beck Depression Inventory score <10, and a STAI-S score of <40). A meta-analysis (
Davidson and Parker, 2001) encompassing 34 studies of EMDR in a variety of populations, including 20 studies of PTSD or treatment of a trauma memory, concluded that while EMDR is superior to no treatment and to non-exposure therapies, it is no more effective than other exposure therapies, and that the eye movements appear to be unimportant in its overall efficacy.
Does combining medication treatment with exposure therapy enhance treatment outcomes? In a study by Rothabuam et al. (unpubished observation), 64 patients who had been treated with sertraline for 10 weeks were randomized to continue sertraline alone or to receive, in addition, PE for 10 sessions over 5 weeks. In the entire sample, the effect size for the patients who received sertraline treatment alone was 1.7, while that for the combination was a striking 2.8. There was evidence of a floor effect; adding exposure therapy greatly improved treatment outcomes for patients with partial responses to medication, but not for those with excellent responses. However, in a subsample of 45 patients, those who had received PE did not relapse on discontinuation of medication, whereas 30% of those who had not had PE relapsed (
Cahill et al., 2003).
The techniques of PE readily lend themselves to dissemination in order to serve a larger population. In a recent study by Foa et al. (in press), Masters-degree level therapists with no expertise in CBT were trained and supervised in PE with and without cognitive restructuring. Training consisted of 5 days for PE and 5 days for cognitive restructuring and 2 h of weekly supervision. These counselors went on to successfully treat patients with long-standing PTSD (mean duration of 9 years), some of whom had been refractory to other treatments. The initial reluctance of the trainees to make patients “suffer” from the exposure techniques was replaced by adoption of the treatment after experiencing its positive results. Prolonged exposure for PTSD has been selected by the Substance Abuse and Mental Health Services Administration (SAMHSA) as a model program for national dissemination.