Huntington's disease (HD) is a progressive neurodegenerative disorder that is inherited in an autosomal dominant fashion. It occurs in approximately 3 to 8 per 100,000 population in the United States. HD has been reported in all races and cultures, but it occurs less in individuals of African or Japanese descent. HD was first formally described by George Huntington in 1872 and has been studied as a classic neurodegenerative disorder since that time. An excellent recent review summarizes historical, diagnostic, and clinical aspects of the disease.
1 Folstein's text provides a more comprehensive treatment of this subject.
2 In brief, HD presents with either an adult or a juvenile onset. The adult form is more common, with onset between 35 and 45 years of age and progression to death within 15 to 20 years. The juvenile form (usually paternally transmitted) presents before age 20 and has a more rapid progression. Although most clinicians are aware of the classic motor findings in HD (involuntary choreiform movements, dysarthrias, dystonias, and rigidity), the psychiatric and cognitive findings, almost always present, are often overlooked.
All patients with HD develop a progressive subcortical dementia that is characterized by frontal lobe executive dysfunction and memory deficits. Neuropsychological testing reveals deficits in recent and remote memory; impaired visuospatial function; difficulty with shifting sets, planning, and organization; and overall decreasing IQ. Psychiatric syndromes (present in up to 79% of patients) most commonly include impulse control disorders, depression, personality changes, and, more rarely, psychosis or mania. Symptoms include disinhibition, irritability, aggression, apathy, and neurovegetative markers of depression. The suicide rate has been reported to be up to 20 times that of the general population over age 50.
1 Increased criminal behavior and hypersexuality have often been reported in HD patients. A recent Danish study found that there is a statistically significant increase in nonviolent crime in male HD patients compared with their nonaffected relatives and control subjects.
3 These criminal behaviors, as well as the psychiatric syndromes, are felt to be related to the destruction of the medial caudate, which disrupts the memory and emotion tracts descending from the frontal lobes.
1,4In 1993, the Huntington's Disease Collaborative Research Group discovered the genetic defect responsible for HD. It is a long repetition of the trinucleotide CAG (cytosine-adenosine-guanosine) sequence in the first exon of a gene on chromosome 4. This repeating sequence leads to the production of a protein called huntingtin. Accumulation of this protein is theorized to lead to nuclear inclusions and cell death in medium spiny GABAergic neurons of the caudate.
5 Currently, probability studies are under way to coordinate the number of CAG repeats with expected age at onset, symptoms, and length of survival.
6Several methods of studying brain functioning, including magnetic resonance spectroscopy (MRS), single-photon emission computed tomography (SPECT), and positron emission tomography (PET) have been applied to HD. MRS provides a relative measure of certain brain metabolites, most commonly presented as the spectrum of the amount of signal produced by the metabolites being measured rather than as an image. MRS uses the same scanners and magnets as traditional MR imaging, but with special hardware and software that allow substances other than water to be studied. Using
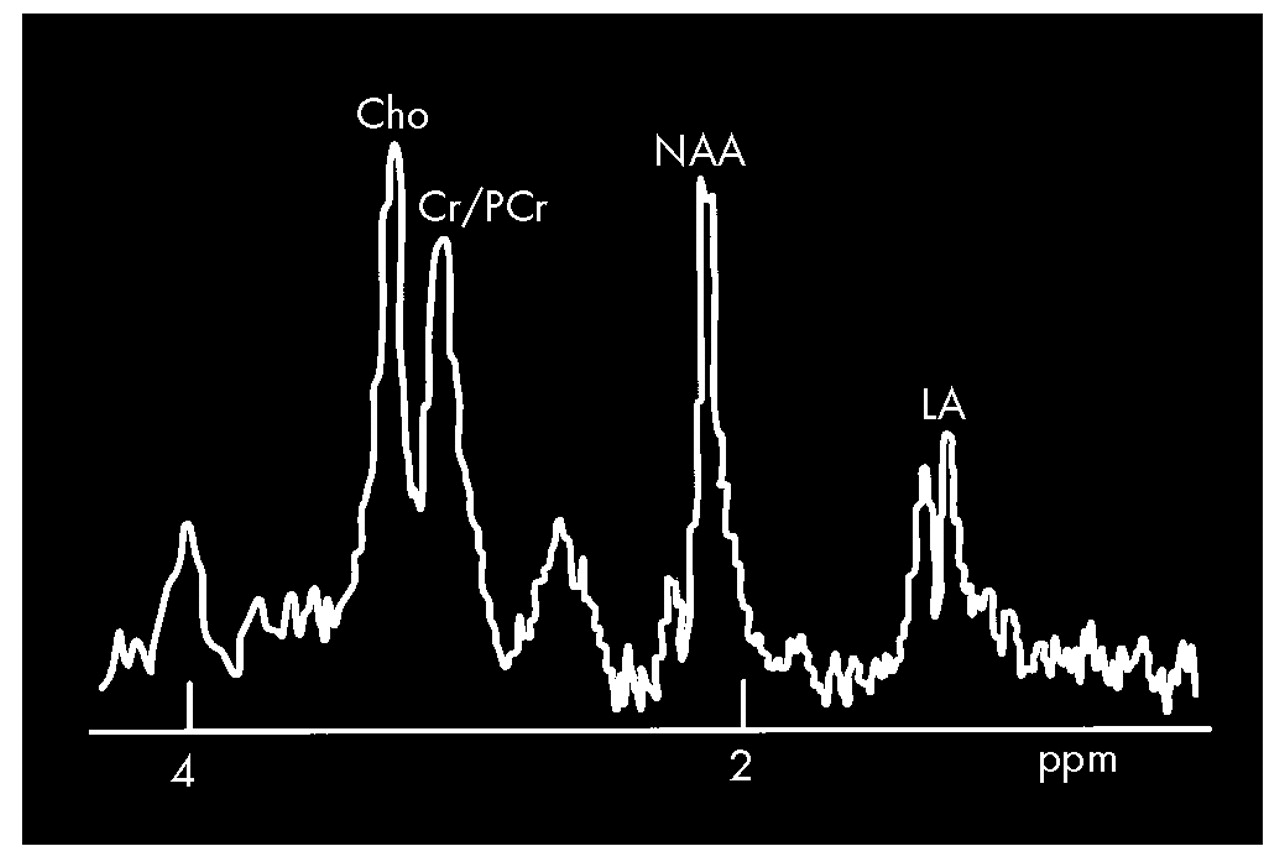
1H (proton) MRS, several groups have found decreased levels of
N-acetylaspartate (NAA, a neuronal marker) in the cerebrum of symptomatic gene-positive patients.
7–12 This change was particularly common in the occipital cortex and striatum.
7,9,11,12 Some have also reported increased levels of lactate (LA; an indication of metabolic distress), although this is still controversial.
13 Increased LA was asymmetric in the striatum, with levels in the left hemisphere exceeding levels in the right.
7 The degree of decrease in NAA and increase in LA in the striatum has been shown to correlate with the duration of symptoms.
7 Experimental treatment with coenzyme Q10 resulted in significant decreases in cortical LA in symptomatic patients,
11 indicating a possible defect in energy metabolism. Asymptomatic carriers typically are characterized by normal
1H MRS spectra, although elevated LA has been reported in some.
7,8 A study using
31P (phosphorus) MRS found a decrease in the phosphocreatine (PCR) to inorganic phosphate (P
i) ratio in the resting calf muscle of symptomatic patients, another indication of a defect in energy metabolism.
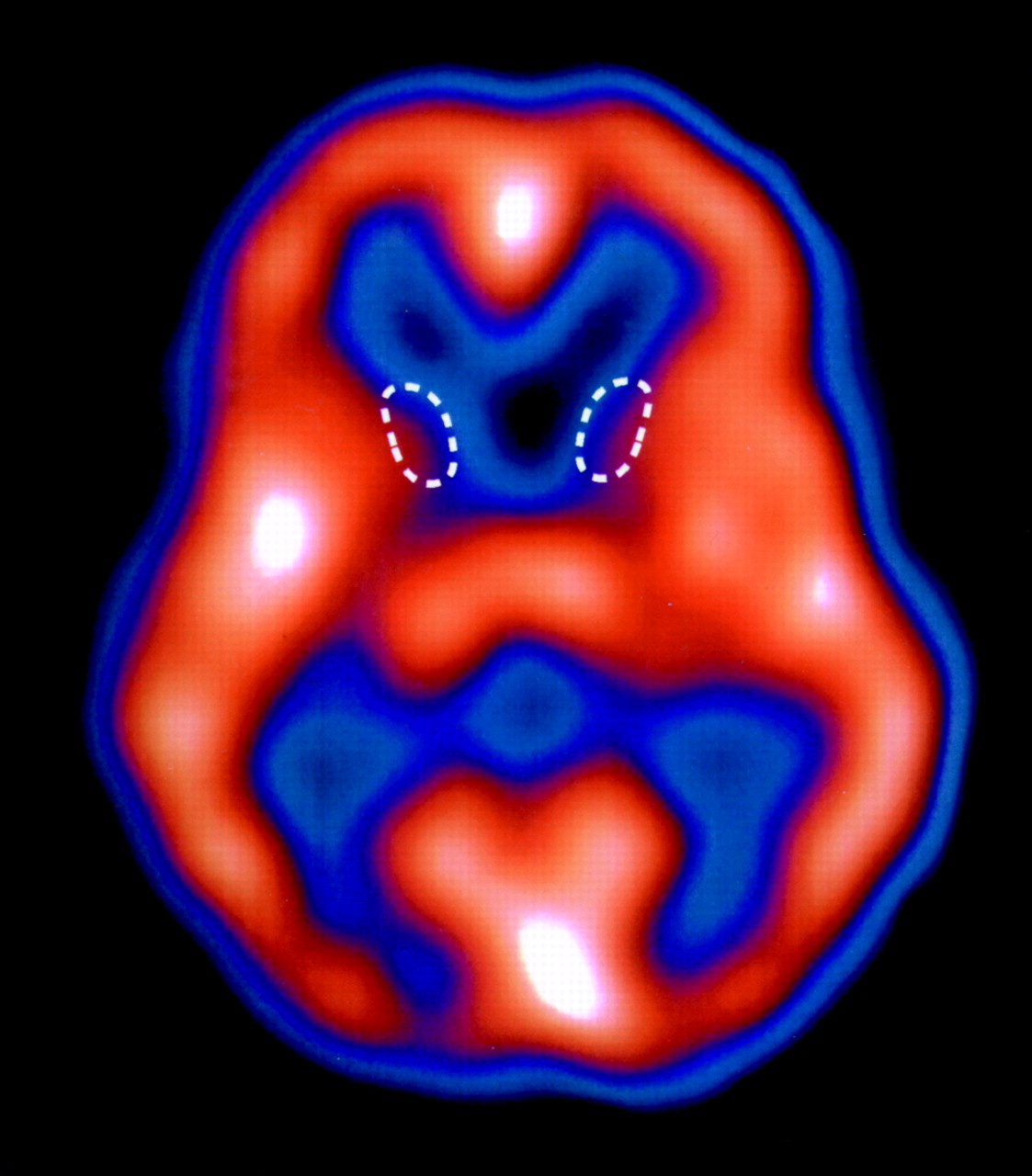
In both SPECT and PET, radioactive tracers are used to measure quantities such as cerebral blood flow, glucose metabolism, and receptor density. It has been known since the 1980s that these methods can demonstrate reductions in caudate glucose metabolism (by PET scanning) and caudate blood flow (by PET and SPECT scanning) in patients with HD prior to clear evidence of structural changes on magnetic resonance imaging (MRI) or computed tomography (CT). More recent studies have suggested that reduced putamen volume can also be seen quite early in the disease by using MRI.
14 Cortical damage can also be demonstrated earlier in the course of the disease on SPECT or PET than on MRI, as is the case with other dementias, such as Alzheimer's disease.
15 There is a good correlation between the reductions in striatal and cortical blood flow and the degree and type of neuropsychological impairment, indicating that SPECT scans may be useful in assessing the degree of neuronal damage and disease progression.
16 PET scanning with radiolabeled neuroreceptor ligands, such as the D
2 ligand carbon-11 raclopride, has revealed markedly reduced dopamine receptor density in the striatum of HD patients.
17 D
2 radioligands for SPECT imaging have shown similar results and may soon be available for routine clinical use. These results are especially interesting clinically, given that SPECT, unlike PET, is now available at nearly all medical centers.
The role that these new approaches to HD will play in clinical management is not yet clear. However, the results obtained thus far with functional brain imaging demonstrate the potential usefulness of these modalities for evaluating both asymptomatic and symptomatic HD patients, particularly in monitoring both disease progression and the effects of therapy.
To date, there is no treatment for Huntington's disease. Typical antipsychotics such as fluphenazine and haloperidol have been used to decrease the choreiform movements early in the course of the disease, but they produce significant side effects, including tardive dyskinesia and worsening cognition. There are limited reports of improvements with both clozapine and electroconvulsive therapy. However, the most innovative and potentially useful treatment is neuronal cell transplantation via stereotaxic injection.
18 This method is ethically controversial because embryonic donors provide the only viable source of stem cells for this therapy. Extensive research is currently under way to develop new strategies to grow early stem cells in the laboratory.
The techniques discussed here are certainly exciting and are leading to a new understanding of Huntington's disease and possible treatments. The potential now exists to apply these concepts to other neurodegenerative diseases.