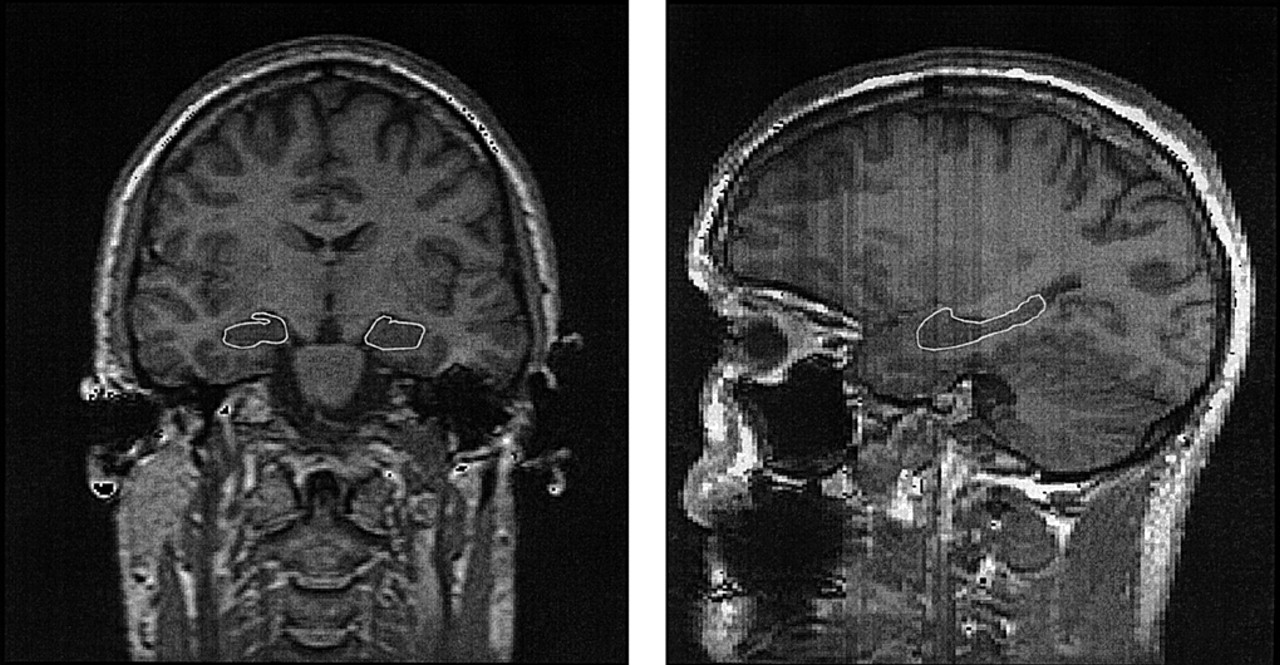
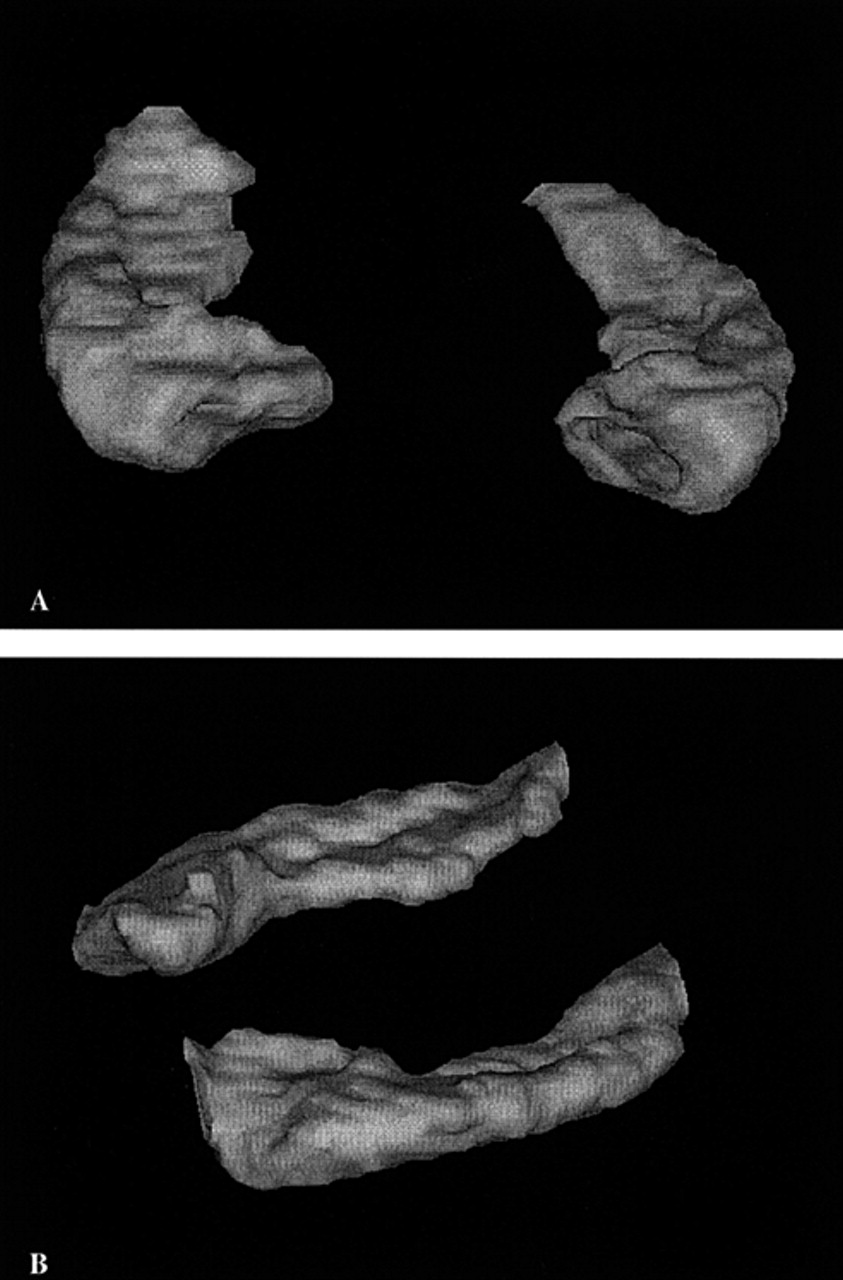
Traumatic brain injury (TBI), at all levels of initial severity, may produce persistent neuropsychiatric symptoms,
1–3 including impairment of auditory gating.
4,5 Auditory gating is a pre-attentive cognitive function that facilitates selective attention by providing a mechanism for filtering out (gating) irrelevant or excessive auditory stimuli.
6 It is largely dependent on the ability of cholinergic afferents from basal forebrain cholinergic nuclei (Ch1 and Ch2)
6–10 to effect multimodal sensory (including auditory) gating within the hippocampus. Cholinergic projections to the hippocampus are particularly vulnerable to disruption by TBI.
4,11–13 Additionally, hippocampal cortex may be particularly vulnerable to direct biomechanical injury during TBI.
14,15 Together, these factors may combine to produce impairment of hippocampal functioning after TBI, the consequences of which include impairments in auditory gating.
Auditory gating may be indexed by the P50 evoked response to paired auditory stimuli.
6,7,16 The amplitude of the P50 evoked responses to these paired auditory stimuli (which consist of a conditioning and a test click) are used to construct a ratio (P50 ratio) of the ability to inhibit, or gate, auditory cortical responses.
8,10 When the P50 ratio is abnormally high (P50 nonsuppression), it suggests that the hippocampus is unable to adequately inhibit responses to repetitive auditory stimuli. Clinically, this form of impaired inhibition is manifest as
impaired auditory gating, a condition in which patients are unable to filter out irrelevant auditory stimuli and consequently fail to consistently mount robust selective attention.
We recently investigated the relationship between TBI, impaired auditory gating, and P50 suppression and demonstrated persistent P50 nonsuppression among TBI patients with symptoms of impaired auditory gating.
5 We interpreted this finding as evidence of hippocampal cholinergic dysfunction following TBI, consistent with our previous speculation
4 and echoing similar suggestions by other authors.
11–13,17,18 These suggestions have been referred to as the
cholinergic hypothesis of attention and memory impairment following TBI.
4 This hypothesis suggests that cholinergic dysfunction may be produced by even relatively mild degrees of concussive injury and that persistent symptoms of impaired auditory gating following TBI may be understood, at least in part, as a consequence of hippocampal cholinergic dysfunction.
More immediately relevant to the cholinergic hypothesis, Waldo et al.
20 demonstrated an association between P50 nonsuppression and reduced hippocampal volume on MRI among patients with schizophrenia. P50 nonsuppression in schizophrenia is believed to reflect hippocampal cholinergic dysfunction due to a genetically determined postsynaptic defect in the alpha-7 nicotinic receptor of hippocampal GABA interneurons
6,7—a fundamentally different mechanism from the cholinergic deafferentation and/or cortical injury that we suggest is the basis for this finding following TBI. Nonetheless, their observation has been used to offer support for the suggestion that P50 nonsuppression is predicated on hippocampal abnormalities.
RESULTS
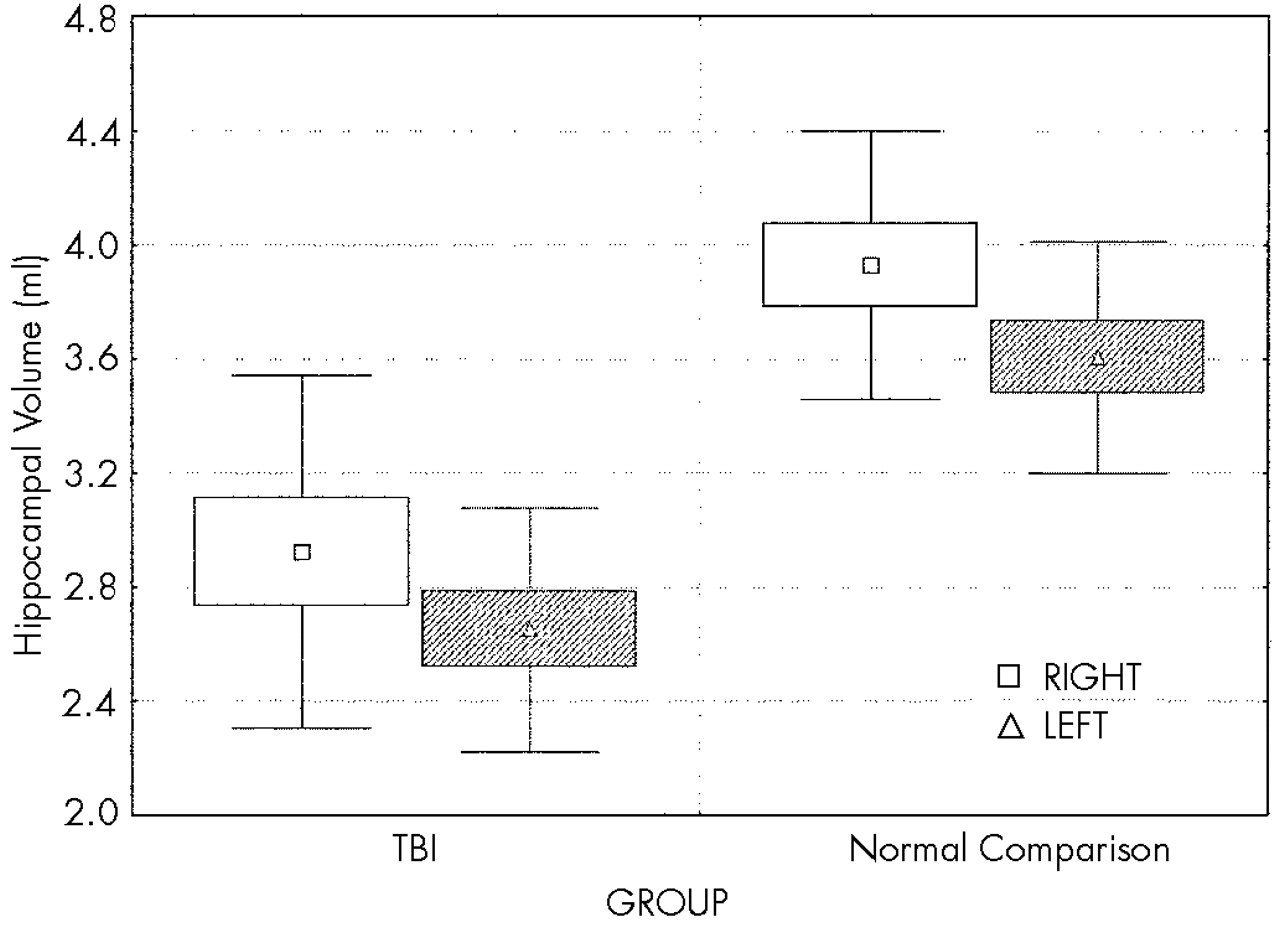
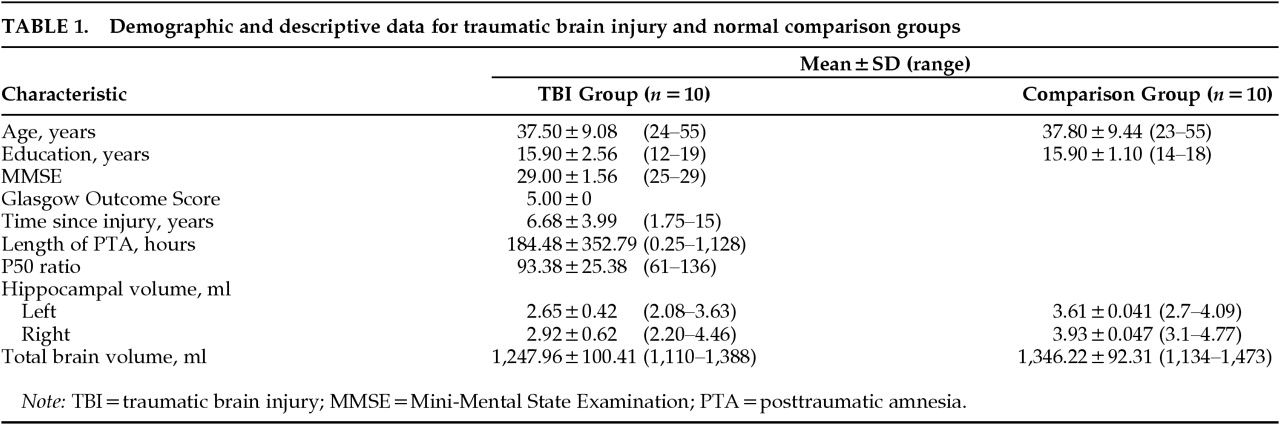
Two-way ANCOVA demonstrated a highly significant effect of group on hippocampal volume (
F=11.9, df=1,17,
P<0.003;
Figure 3), with reduction of hippocampal volume in the TBI group. Planned comparisons demonstrated that both right and left hippocampal volumes in the TBI group are highly significantly less than those of the normal comparison group (
F=8.6, df=1,17,
P<0.009, and
F=15.6, df=1,17,
P<0.001, respectively). Although there was an effect of group on total brain volume (smaller total brain volume in the TBI group,
F=5.2, df=1,18,
P<0.04), this effect did not diminish the significance of the effect of group on hippocampal volume. There was a significant hemisphere effect on hippocampal volume in both groups (
F=28.4, df=1,16,
P<0.001), left hippocampus being smaller than the right, but there was no interaction between group (TBI vs. comparison) and hemisphere, suggesting that the volume loss in the TBI group is relatively symmetric.
Duration of posttraumatic amnesia, taken as an indication of TBI severity, was not significantly correlated with total brain volume (r2=0.19, P=0.20), left hippocampal volume (r2=0.00, P=0.99), or right hippocampal volume (r2=0.00, P=0.89).
DISCUSSION
The present study was performed to investigate whether our hypothesis of hippocampal cholinergic dysfunction following TBI could be further supported with evidence of structural hippocampal abnormalities in TBI patients with symptoms of impaired auditory gating and P50 nonsuppression. The results of this study suggest a disproportionate effect of TBI on the hippocampus, with volume reduction in this structure being greater than can be accounted for by reduction in total brain volume. Disproportionate hippocampal volume loss among P50-nonsuppressing TBI patients provides support for the cholinergic hypothesis of auditory gating impairment following TBI by demonstrating convergent structural and electrophysiologic evidence of hippocampal abnormalities in these patients. Although the sample size in this study is relatively small, the effect size (ω
2=0.31) derived from the data acquired suggests that this sample is large enough to permit a meaningful investigation of differences in hippocampal volume between these two groups, with power between 0.8 and 0.9.
31 Additionally, the significance value of effect of group on hippocampal volume (
P<0.003) suggests that the likelihood of a Type I statistical error is very low. Taken together, these analyses offer some reassurance that the findings may be regarded as robust even in the context of a relatively small study. To make the potential implications of these findings as clear as possible, we provide below a brief review of the cholinergic hypothesis.
The cholinergic hypothesis of attention and memory impairment due to TBI is based on the premise that selective and sustained attention derive from the function of and relationships among a selective distributed network of central nervous system structures.
32 The network includes the reticular formation,
33–35 thalamus,
36,37 hippocampus and entorhinal areas,
32,38 bifrontal and right parietal lobes,
32,39 and axonal connections between these structures.
32,40 Within this network, the hippocampus is required for sensory gating, the pre-attentive cortical process that facilitates the filtering of relevant highly processed multimodal sensory information from irrelevant background stimuli and the presentation of relevant information to the network for attentional and additional processing.
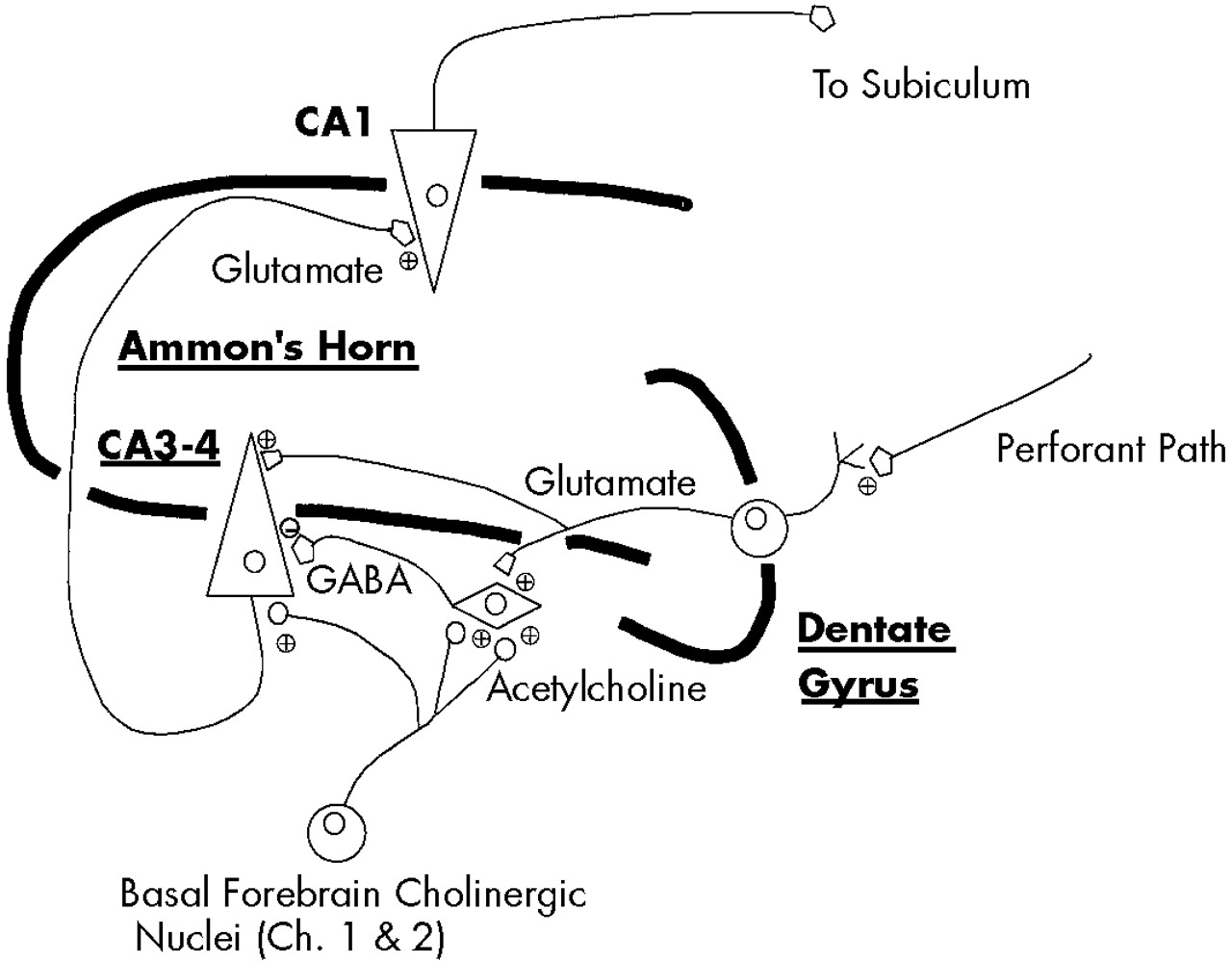
8,32The flow of highly processed sensory information through the hippocampus begins with its entry into the dentate gyrus and CA3 region, from whence it flows to the CA1 region and then to other subcortical and neocortical sites
16,32,41,42 (
Figure 4). As information enters the CA3 region it is directed not only through the direct afferent pathway to glutamatergic neurons, but also through a recurrent pathway that terminates on CA3–CA4 GABA interneurons. Activation of these GABA interneurons results in transmission of inhibitory signals to the CA3 glutamatergic neurons, momentarily preventing additional flow of sensory information through the direct pathway. Acetylcholine, via its action at low-affinity alpha-7 nicotinic receptors,
43 provides an excitatory postsynaptic potential (EPSP) to these CA3–CA4 GABA interneurons, permitting their activation upon receipt of information from the recurrent pathway.
6,7,16,16,44 Without cholinergic input to provide this EPSP, the GABA interneurons are not fully responsive to stimulation by incoming information from the recurrent pathway and therefore cannot provide adequate inhibition of subsequent incoming sensory input in the direct pathway. The cholinergically dependent momentary interruption of the flow of information through the hippocampus provided by these CA3-CA4 GABA interneurons is a principal means by which sensory filtering, or sensory gating, occurs.
Cholinergic afferents from the basal forebrain cholinergic nuclei (Ch1 and Ch2) project to the CA3-CA4 GABA interneurons in the hippocampus and appear to be particularly vulnerable to injury from even mild to moderate concussive forces.
11–13,18,45–48 TBI may disrupt these cholinergic afferents through direct mechanical trauma,
14,49–51 diffuse axonal injury,
14,52–55 and/or disturbance of cholinergic function within the hippocampus.
11,38,45,47 When cholinergic function in the hippocampus is disrupted because of TBI, impairments of auditory sensory gating and attention may develop and may be electrophysiologically indexed by nonsuppression of the P50 evoked response to paired auditory stimuli.
4,5Given this background information, the cholinergic hypothesis of attention and memory impairment following TBI states that 1) selective and sustained attention by the distributed attentional network are prerequisites for normal declarative/episodic memory; 2) pre-attentive filtering of highly processed sensory (including auditory) stimuli by the hippocampus is a prerequisite for the development of normal selective attention; 3) the process of auditory gating in the hippocampus is cholinergically dependent; 4) TBI disrupts hippocampal cholinergic function; and 5) therefore, TBI may impair attention and memory via disruption of the cholinergically dependent process of auditory gating.
4,5The patients studied here have symptoms of impaired auditory gating and P50 nonsuppression, both of which are consistent with the hypothesis of impaired hippocampal cholinergic function following TBI. The disproportionate hippocampal volume loss observed in these patients further supports this hypothesis by offering neuroimaging evidence of a structural hippocampal abnormality that converges with the symptoms and abnormal P50 suppression of these TBI patients. The hippocampal atrophy observed in these TBI patients most likely reflects a combination of deafferentation due to diffuse axonal injury and some degree of both direct and secondary cortical injury. These are predictable consequences of the biomechanical forces experienced during TBI.
14,18,46,56,57 We suspect that diffuse axonal injury may contribute more strongly to the present finding. Reduced hippocampal volume measurable on MRI may reflect reduced white matter volume due to axonal losses. Although there may be secondary reduction in the number of hippocampal neurons following Wallerian degeneration of injured axons back to their somae in the hippocampus, the neurocircuitry described in
Figure 4 suggests that injury to cholinergic fibers would more likely result in cell losses in the basal forebrain cholinergic nuclei than in the hippocampus itself. This interpretation is best supported by the ability of these patients to generate a P50 evoked potential (which requires hippocampal cortex) of reasonable amplitude despite hippocampal volume loss.
However, this study cannot clarify the relative contributions of cortical loss and white matter loss to the hippocampal volume loss observed here, so this interpretation must remain speculative at present. Additionally, and perhaps more troubling, this study cannot definitively determine whether the small hippocampal volume of these TBI patients is an effect of injury or is instead a premorbid risk factor that contributes to the development of auditory gating, attention, and memory impairments following TBI. Although the cholinergic hypothesis discussed herein suggests that the former possibility (hippocampal volume loss as an effect of injury) is more likely, additional studies are needed to further clarify this issue. These studies should employ methods such as proton magnetic resonance spectroscopy (e.g., using the NAA:Cr ratio to assess whether the hippocampus is small but composed of viable neurons or whether there is spectroscopic evidence of neuronal injury) and/or prospective (pre- and post-TBI) measurement of hippocampal volume. Further, information on the relationship between the present findings and attention and memory cannot be provided here because neuropsychological examination results relevant to this question are lacking. Further studies are needed to clarify the existence, nature, and strength of any such relationships.
Another interesting, although preliminary, result of this study is the finding of a relatively similar degree of hippocampal volume loss among all the TBI patients despite disparate initial TBI severity. It is important to restate that the TBI subjects in this study were included only if they had experienced relatively good global clinical outcome, had not developed cognitive impairments severe enough to warrant a dementia diagnosis, and generally appeared to be the sort of patients often described as the “walking wounded,” regardless of initial TBI severity. The intent of this sample design was to enable us to evaluate patients with respect to their long-term clinical outcome from TBI and to begin to address whether there are differences in hippocampal volume between persistently symptomatic, P50-nonsuppressing TBI patients with initially mild/moderate versus those with severe injuries. Using this same sample design in our previous study of P50 suppression, we found no significant differences between 11 mild/moderate initial TBI and 9 severe initial TBI subjects, although the P50 ratios of all TBI subjects were highly significantly different from those of the normal comparison subjects.
5 In other words, it appeared that merely having persistent symptoms of impaired auditory gating was a more robust indicator of the presence of this electrophysiologic abnormality than one would anticipate based on the initial injury severity alone.
The present results also appear to provide preliminary support for the idea that even in apparently “mildly” affected TBI patients (as judged by GOS and MMSE), the presence of symptoms that are referable to a specific underlying neurophysiology is a stronger indicator of the presence of structural brain abnormalities than would be expected based on the initial severity of TBI alone. However, a negative finding in a sample of this size does merit concern that we may lack adequate power to detect correlations, if they exist, between initial TBI severity (as assessed by duration of PTA) and the volumetric variables. These findings nonetheless do suggest that additional structural and/or functional neuroimaging and electrophysiologic studies of these “good outcome” but persistently symptomatic TBI patients are important to pursue further.