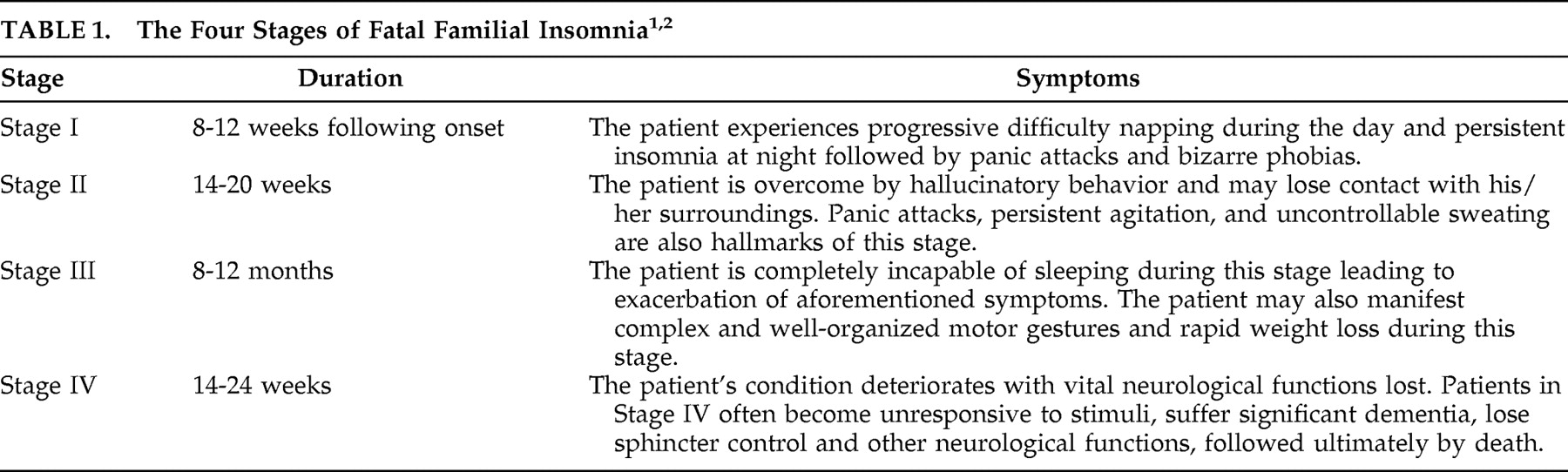
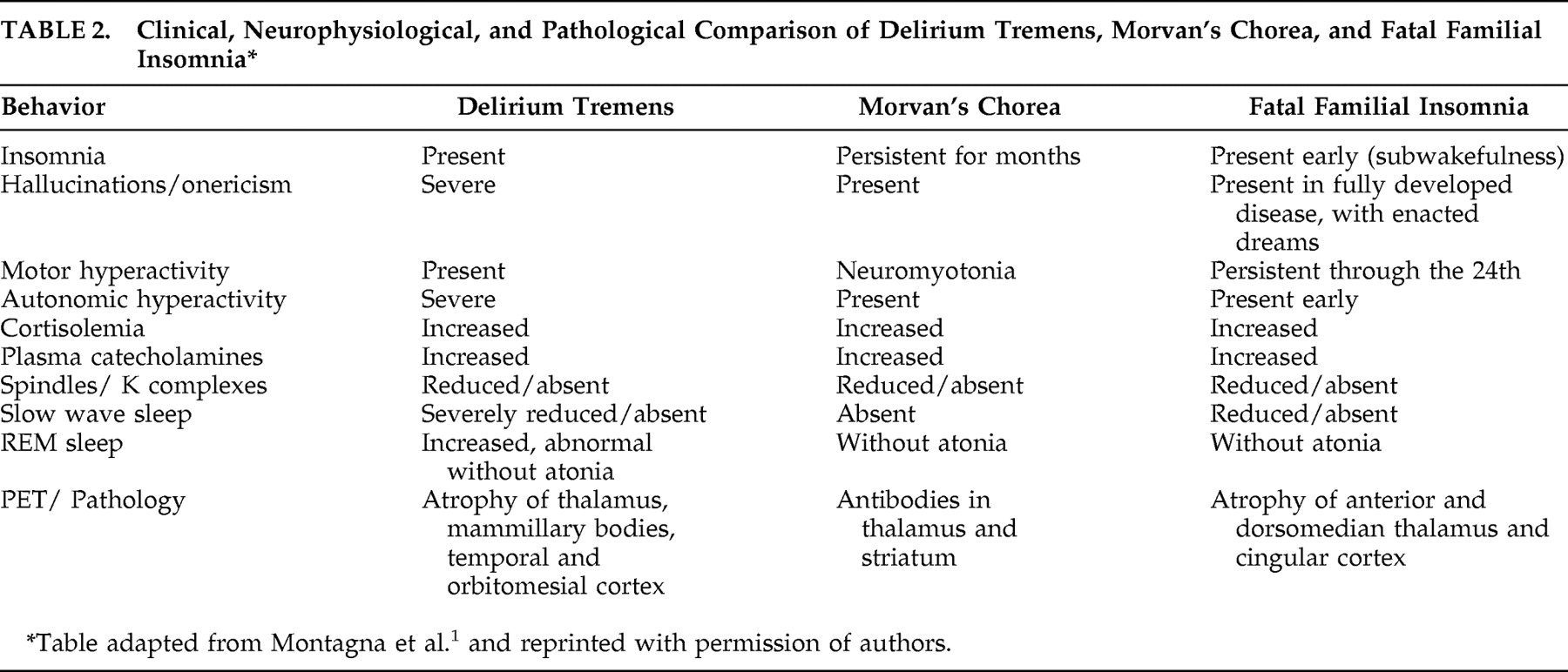
Morvan’s chorea, or “fibrillary chorea,” describes the rare condition in which neuromyotonia (involuntary contraction of muscles at rest) occurs in the setting of peripheral nervous system hyperexcitibility.
1,
3,
20 The typical movements in Morvan’s chorea most commonly affect bilateral calves and the posterior part of the thigh. The peripheral nerve hyperexcitibility that dominates in Morvan’s chorea typically results in neuromyotonia as well as hyperhydrosis, paresthesias, dysautonomia, myalgias, incontinence, palpitations, and sleep dysfunction.
3,
20,
21 Similar to the other two components of agrypnia excitata, Morvan’s chorea also shares the same clinical features as both FFI and delirium tremens: agrypnia, mental confusion, and a significant reduction of slow wave sleep (
Table 2 ).
1 –
3,
18 Unlike the other components of agrypnia excitata, however, Morvan’s chorea can arise at any age and may occur with painful cramps.
1,
21 Like patients with FFI, Morvan’s chorea also may manifest with hallucinatory behavior, a stuporous confused state, and peculiar motor disturbances, but these features are very rare in Morvan’s chorea.
1,
22 Like the other subtypes of agrypnia excitata, polysomnograms in patients with Morvan’s chorea reveal the persistent absence of sleep rhythms for up to 4 months with the typical loss of slow-wave sleep.
1,
21 Patients with Morvan’s chorea will exhibit REM without atonia and will also demonstrate hypercortisolemia and hypercatecholemia in the plasma.
2,
21The exact pathophysiology of Morvan’s chorea remains uncertain. However, it is believed that the condition arises via autoimmune mechanisms.
21 The presence of voltage-gated potassium channel antibodies is also commonly found in the serum of patients with Morvan’s chorea.
20,
21,
23 The effect of these antibodies is believed to cause neuronal hyperexcitability by disrupting the ability of voltage gated potassium channel antibody to repolarize motor nerves effectively.
21,
24Unlike FFI, which runs a uniformly fatal course, Morvan’s chorea may resolve overtime but can, in some cases, result in death.
1,
2 In severe cases encephalopathy, dementia, or reduplicative paramnesia may ensue.
2,
20,
23 Notwithstanding, given the potentially fatal course of other diseases that mimic Morvan’s chorea, consideration of a thorough differential diagnosis is critical to help minimize misdiagnosis.
25 Accurately recognizing Morvan’s chorea may help prevent misdiagnosis in the setting of clinically similar diseases such as in patients suffering from acquired forms of peripheral nerve hyperexcitability. Acquired peripheral nerve hyperexcitability can occur following timber rattlesnake envenomation or Guillain-Barre’ syndrome, two conditions that can lead to devastating sequelae.
22,
25 Fortunately, Morvan’s chorea is, for the most part a treatable condition.
1,
2,
3 Unlike the other two components of agrypnia excitata, appropriate therapeutic approaches can reverse the course of Morvan’s chorea.
26 Plasmapheresis, thymectomy, and long-term immunosuppressive therapy can yield complete resolution of symptoms in patients with Morvan’s chorea.
20,
26