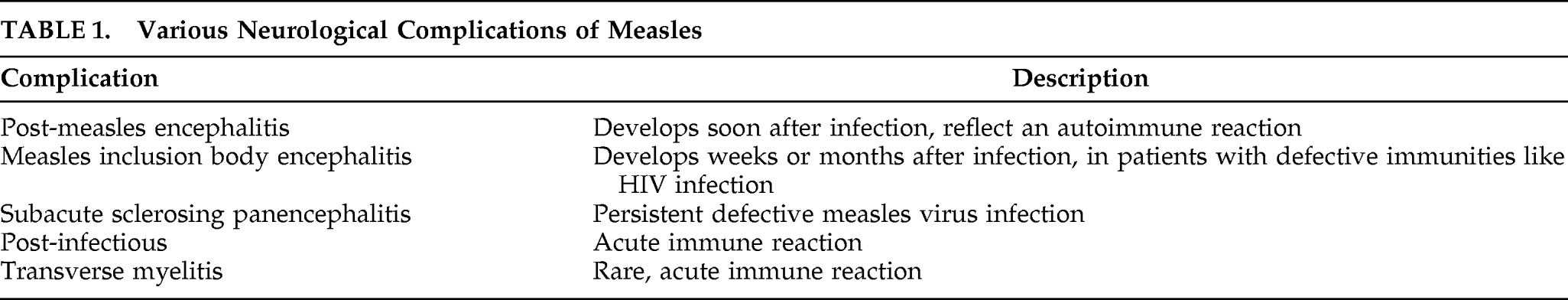
To the Editor: Subacute sclerosing panencephalitis (SSPE) is a serious disorder of the CNS. It is a slow virus infection caused by defective measles virus (
Table 1).
The term subacute sclerosing panencephalitis has been used since Greenfield suggested it in 1960 to designate a condition due to a persistent infection by a virus involving both gray matter and white matter.
1 In fact, SSPE had originally been described as three different neuropathological entities. In 1933, Dawson
2, for the first time, described a child with progressive mental deterioration and involuntary movements who, at necropsy, was found to have a dominant involvement of gray matter in which neuronal inclusion bodies were abundant. He suggested the term “subacute inclusion body encephalitis.” Later Pette and Doring
3 reported a single case of what they called “nodular panencephalitis” a disease with equally severe lesions in both gray and white matter. Six years later, Van Bogaert
4 drew attention to the presence of dominant demyelination and glial proliferation in the white matter and suggested the term “subacute sclerosing leukoencephalitis.” A viral etiology was suggested by Dawson, but it was Bouteille et al.
5 in 1965 who demonstrated the presence of viral structures resembling measles virus in the brain on electron microscopy. In 1969, the measles virus was actually recovered from the brain of a patient with SSPE.
6The annual incidence rate varies from one to four per million people in developed countries.
7 However, in developing countries it is high and quite variable due to a higher proportion of the total population that is younger than 2 years of age, a higher proportion of measles cases in the very young, more frequent subclinical measles virus infection, and perhaps other host factors.
8 In India, the annual incidence rate is reported to be as high as 21 per million people.
7,8 The latent period between measles infection and SSPE is around 6–8 years in most of the cases, but may range between 3 months to 18 years.
Initial presenting symptoms have commonly been reported to be myoclonic jerks and falling attacks, changing gait, abnormal movements, speech impairment, inability to walk or stand, seizures, dementia, visual disturbances, pyramidal and extrapyramidal signs.
Behavioral disturbances have also been reported as common initial presentation. However, cases where catatonia has been the initial presenting complaints are rare. We wish to report a 13-year-old boy who initially presented with catatonic symptoms only and was later on diagnosed as a case of SSPE based on typical EEG and CSF findings.
Case Report
A-13-year old Hindu boy in the seventh grade, a product of non-consanguineous marriage, presented with an illness of insidious onset, a progressive course of 5 months duration with no apparent precipitating factor, and complaints of withdrawn behavior and decreased interaction with others. Over a period of around 2 months, he gradually stopped speaking completely and would not even ask for food. He also started standing for long periods of time and would also resist movements. He would not obey any commands given to him. He needed to be force fed and on occasion would clench his teeth so that it would be difficult for the family members to feed him. On occasion, after about 3 months from the start of illness, our patient would start abusing his family members and would even get physically violent for no apparent reason. There was no history of any prolonged high-grade fever or history of abnormal body jerky movements or history of fits.
The patient had stopped going to school during this period. His sleep was also decreased because he would often keep standing for long periods of time during the night. He would not take care of himself and would have to be forcefully bathed. Seeing this condition, family members initially consulted faith healers. Seeing no improvement after about 4.5 months from the onset of illness, the parents brought boy to a doctor, who prescribed risperidone, 2 mg/day, and lorazepam, 4 mg/day. The patient continued the treatment for 2 days when he suddenly developed sustained prolonged contraction of the neck muscle (suggestive of dystonia) which was relieved by an injection of promethazine. Because of this, the patient was referred to us, where he was admitted for a detailed assessment.
There was no history to suggest motor, sensory, and bladder or bowel involvement. There was no history of bleeding diathesis, cardiac disorder, or exposure to tuberculosis. He had no family history of any psychiatric illness. His early birth and developmental history were not significant. However, he was not immunized against measles and had suffered from measles at 11 years old. He had two elder male siblings and one younger brother, all of whom were healthy.
On examination at admission, he was conscious, afebrile, and his vitals were stable. He was not talking at all. The boy would keep standing for long periods of time and would resist any movements. He would not follow the commands given to him and would have lead pipe rigidity in both the upper limbs. No other abnormalities were found upon neurological examination (though a detailed examination was not possible as he was not cooperative). Fundus examination was bilaterally normal. Investigations revealed hemoglobin of 12.5g/dl, total leukocyte 9,000/mm3 with 62% polymorphs, an erythrocyte sedimentation rate of 20 mm in the first hour and normal urine and stool examination. Blood tests for toxoplasma, cytomegalovirus, and herpes simplex virus IgG and IgM were negative. His venereal disease research laboratories (VDRL) slide test was negative. Chest X-ray was normal and mantoux was negative. CSF was grossly clear with normal opening pressure and showed 5 lymphocytes/ mm3, protein 20 mg/dl, and sugar 70 mg/dl against a blood glucose of 100 mg/dl.
The Bush Catatonia Rating Scale was applied, on which the patient scored 7. A provisional diagnosis of catatonia was kept and we began treatment with lorazepam, gradually increased to 8 mg/day. He did not show any improvement with this treatment but instead he deteriorated. He became drowsy. About 4 days after admission, the patient developed sudden abnormal jerky movements of the upper limbs during the night time suggestive of myoclonus. In view of this finding, an EEG and a brain MRI were done. EEG was characterized by periodic complexes consisting of bilaterally symmetrical, synchronous, high-voltage (200–500 mv) bursts of polyphasic, stereotyped delta waves. The EEG was characteristic of SSPE. MRI of the brain revealed multiple T2 hyperintense foci in supratentorial subcortical white matter (possible post-encephalitis demyelinization). Furthermore, his CSF was sent for serology and was positive for IgG anti-measles antibodies. A diagnosis of subacute sclerosing panencephalitis was made, and the patient was referred to the department of neurology. He was there started on oral sodium valproate and injected interferon. The prognosis was explained to the parents. They took the child home against medical advice. The patient was then lost to follow-up.
Discussion
The diagnosis of subacute sclerosing panencephalitis (SSPE) requires the fulfillment of at least three of the five criteria
9 which include the following:
1.
A typical clinical picture: personality and behavioral changes, worsening school performance, followed by myoclonic seizures, paresis, dyspraxias, memory impairment, language difficulties, blindness, and eventually obtundation, stupor and coma;
2.
Characteristic EEG changes
3.
Elevated CSF globulin levels greater than 20% of total CSF protein
4.
Raised titers of measles antibodies in blood and CSF
5.
Typical histopathological finding in brain biopsy or autopsy
However, atypical presentations have been described including isolated psychiatric manifestations, poorly controlled seizures, or isolated extrapyramidal syndromes like chorea, hemiparkinsonism, etc.
10 A stroke-like onset has also been described.
10Initial presentation of catatonia in the literature has been rare. We could find only one case report describing catatonia as the presentation of SSPE.
11 Another report of SSPE found only one case presenting as catatonia among 32 cases studied.
12 In our case, diagnosis of SSPE was confirmed by the typical EEG findings and demonstration of demyelination on MRI along with CSF findings of anti-measles antibodies.
The initial presentation of catatonia in our case is of interest. The patient developed typical features of SSPE-like myoclonus only 5 months after the onset of catatonic symptoms. The patient initially had only symptoms of catatonia without any other symptoms, to the extent that at one point a diagnosis of psychosis was entertained. Our case report demonstrates the importance of ongoing neurological examinations in patients with psychiatric disorders. It also demonstrates the importance of EEG in patients with psychiatric disorders, especially catatonia. It highlights the importance of excluding all possible causes for catatonia especially when only catatonia is a presenting feature without other symptoms of psychiatric disorders. In investigating these patients, EEG remains a simple and cheap modality of investigation and should be considered in all patients with atypical features.
In conclusion, SSPE is a rare complication of a common viral infection. Our case had a conglomeration of atypical and rare features of this rare illness. SSPE is encountered much more commonly in this part of the world than in the west. Therefore, pediatricians, neurologists, and psychiatrists are more likely to come across SSPE in its typical, atypical, and rare presentations. A high index of suspicion is needed to detect SSPE in its atypical and rare forms. When faced with a young patient with psychiatric manifestations, especially catatonia with no other significant history and with history of measles infection in the past, SSPE is always a diagnosis to be excluded.