Since it was first described in 1966 by Brain et al.,
1 Hashimoto's encephalopathy (HE) has gained prominence in the differential diagnosis of encephalopathy of unknown origin. The increased number of cases in the literature demonstrates the growing interest of the scientific community in this condition, which still requires a more thorough understanding of its pathophysiology, diagnosis, and treatment. Hashimoto's encephalopathy (HE) is a rare neuropsychiatric syndrome, more common in women, associated with serologic evidence of antithyroid antibodies, when other causes of encephalopathy are excluded.
2 It may have an acute onset, characterized by episodes of cerebral ischemia, seizure, and psychosis, or it may present as an indolent form with depression, cognitive decline, myoclonus, tremors, and fluctuations in level of consciousness.
2,3 The relationship between HE and Hashimoto's thyroiditis (HT) remains uncertain, since there is no evidence that thyroid autoantibodies react with brain tissue and affect neuronal function, and the level of circulating autoantibodies is not correlated with the severity of neurological manifestations or with the response to treatment. The good response to corticosteroid therapy and its association with other autoimmune diseases point to an inflammatory or immunological dysfunction.
2,3 Despite “Hashimoto's encephalopathy” being the most commonly used designation, some authors advocate a change of nomenclature to “steroid-responsive encephalopathy associated with autoimmune thyroiditis (SREAAT),”
4,5 which, in the face of current evidence, would be a more generic and appropriate term for the condition. Considering the variety of neurologic manifestations that may present and the lack of well-defined diagnostic criteria, the disease appears to be underdiagnosed, which has serious consequences for patients, since it is a potentially treatable condition. In this report, we describe a long-standing case of HE with a good response to immunosuppressive treatment. We also performed a systematic review of the literature of cases reported since 1966.
CASE REPORT
The patient was a 23-year-old woman with a 6-year history of weakness, gait ataxia, and cognitive deficit, characterized by amnesia for recent events, dyscalculia, dysgraphia, inattention, and difficulty in finding words, which prevented her from performing routine activities, such as learning, walking, or swimming alone. she also presented with episodes of myoclonus, tremors, and rigidity of extremities and headache and transient aphasia that usually lasted about 15 minutes, sometimes occurring 3 times a day. During this period, the patient had been undergoing evaluation in departments of neurology, psychiatry, and endocrinology, receiving the diagnosis of conversion disorder and hypothyroidism secondary to Hashimoto's thyroiditis. Since then, she had been on levothyroxine 75 mcg/day, clonazepam 2 mg/day, fluoxetine 20 mg/day, and valproic acid 500 mg/day, without a successful response. MRI of the brain and cervico-thoracic spine, electromyography, and EEG were normal.
Laboratory tests showed negative antinuclear antibody (ANA) and anti-DNA antibody rheumatoid factor: <8mUI/ml (normal: <20mUI/ml), negative anti-Ro/SSA, negative anti-HIV I and II, CK: 57U/L (normal: <70U/liter), C-reactive protein: <6 mg/ml; negative skeletal muscle antiacetylcholine receptor: 0.02 nmol/liter (normal: 0–0.2 nmol/liter). TSH: 4.3 mcUI/ml; FT4: 1.15 ng/dl, T3T: 61.3 ng/dl, and antithyroperoxidase (TPO)>1,000 UI/ml (normal: 0–35 UI/ml). The CSF study showed reactivity for anti-TPO, with no infectious or inflammatory findings. In view of these neurologic symptoms, associated with high titers of antithyroid antibodies and the exclusion of other possible causes of encephalopathy, the patient was diagnosed with HE. We started methylprednisolone 1 g/day for 3 days, which resulted in a significant improvement in her clinical condition after the second day of treatment, and which was maintained with prednisone 1 mg/kg/day and azathioprine 1 mg/kg/day. We developed an index of disease activity (myoclonus, diplopia, dysgraphia, memory and cognition problems, fatigue, drowsiness, and headache), which decreased 80% after 10 days of steroid medication. During that period, there was a more marked improvement in the diplopia, myoclonus, gait ataxia, and dysgraphia, while memory and cognition showed a more modest improvement.
One month after discharge, the patient reported a sustained improvement on all parameters, including memory and cognition, making it possible for her to return to an almost normal routine. progressive reduction in prednisone dose (2.5 mg per week) was then started. in the third month of treatment, the patient showed no symptoms of recurrence. However, she again complained of weakness, which started when she was taking 0.5 mg/kg/day (30 mg/day) of prednisone. The authors then increased the dose of azathioprine to 1.5 mg/kg/day and continued the gradual decline in the prednisone regimen.
METHODS
We performed a systematic review of the literature of all cases of HE that had been published since it was first described. We also analyzed the data available on clinical presentation, laboratory, radiological, electroencephalographic findings, and outcome of patients in order to determine the characteristics and outcomes associated with this condition. We conducted a bibliographical search in the PubMed, Cochrane library and EMBASE systems, using the following keywords: “Hashimoto AND Encephalopathy” and “Graves' Disease AND Encephalopathy.” Included in the review were articles designated “case report” or “series of cases” published in English and describing patients diagnosed with HE, defined by the presence of neuropsychiatric clinical findings associated with the following criteria, excluding other causes of encephalopathy: 1) presence of high levels of antithyroid antibodies in serum or CSF; 2) no alteration in the CSF and/or imaging tests compatible with infectious, vascular, or neoplastic etiology; and 3) a good response to immunosuppressive therapy.
Articles were excluded if duplicated, published in languages other than English, or not available for full analysis, and those that did not contain sufficient information to determine whether the disease met the inclusion criteria above; 113 articles were identified that referred directly to encephalopathy associated with autoimmune thyroid disease, of which 56 were excluded because they met at least one of the exclusion criteria and 5 because they could not be accessed.
RESULTS
A total of 52 articles were included in the review. We found 130 patients who met the criteria for diagnosis of Hashimoto's encephalopathy (HE).
Clinical Features
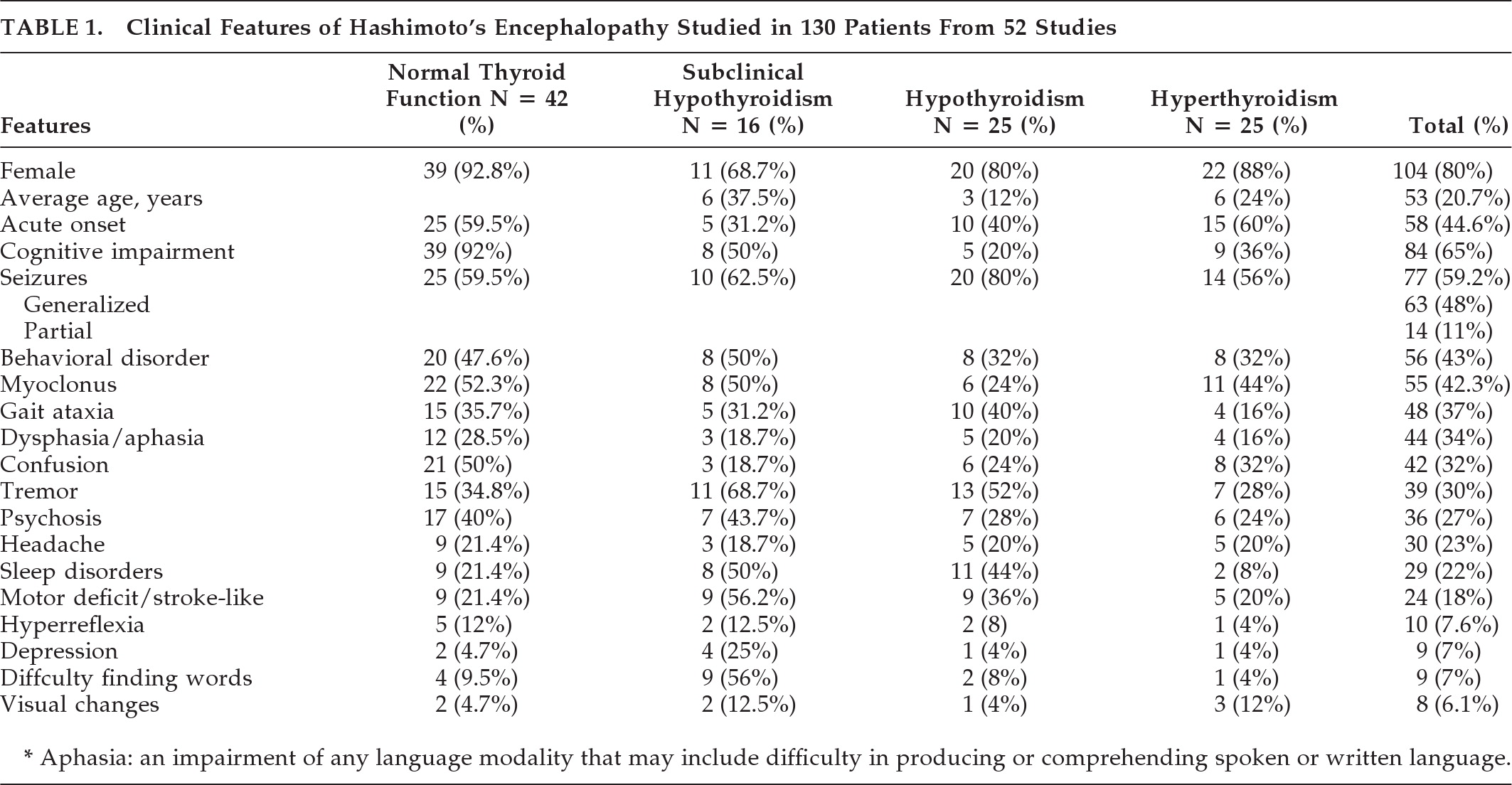
The clinical and laboratory data are summarized in
Table 1.
Thyroid Profile
Most patients were euthyroid at diagnosis (42%/32%), followed by hypothyroidism and hyperthyroidism in equal proportions (25%/19%). Subclinical hypothyroidism was present in 16 cases (12%.) In four case reports, the authors did not report thyroid function. A total of 112 patients (86%) were positive for anti-TPO antibodies, whereas 63 (48%) were positive for antithyroglobulin (anti-Tg). Their levels were not correlated with the severity of the clinical presentation. Antibodies against the amino terminal [propto]-enolase antigen (anti-NAE) were identified in 26 patients, of whom 17 (65% of the sample) were positive.
Cerebrospinal Fluid (CSF)
A high protein concentration was seen as the main feature, which was present in 71.1% of the patients; 20 patients were tested for anti-TPO antibodies in the CSF, of whom 15 (75%) were positive, and 11 (84.6%) of 13 patients tested were positive for anti-Tg. Oligoclonal bands were studied in 21 patients and were present in only 7 (33.3%).
EEG
Of the 118 patients who had EEG data, 105 presented with some type of changes, the most frequent of which was a generalized slowing of waves, present in 71 (59.7%). Other findings included frontal intermittent rhythmic delta activity (FIRDA) and/or delta and theta activity (11 patients: 9.1%), focal and intermittent slow waves (8 patients: 6.7%), epileptiform discharges, and triphasic waves.
Computerized Tomography (CT)
Only 25 patients received a CT scan, and, in 11 of these (44%), some abnormalities were found (Table 4), the main ones being cerebral atrophy (6%–24%) and cerebral infarction (4%–16%).
MRI
Of the 101 patients who underwent MRI scans, abnormalities were seen in 46 (45.5%). Twenty-six scans (25.5%) showed hyper-signal on T2, 6 (6%) cerebral atrophy, 4 (4%) ischemia, and 2 (2%) edema and inflammation.
Treatment and Prognosis
In the cases reported, 62 patients (47.6%) were treated with a regimen of IV methylprednisolone (MPD; 500–1,000 mg/day) for 3 to 5 days, followed by a high initial oral dose of prednisone (1–2 mg/kg/day), after which it was gradually decreased in accordance with the clinical improvement shown; 16 patients (12.3%) received only IV methylprednisolone, 3 of them (3/16) in combination with azathioprine after pulse therapy. In the three cases described, IV dexamethasone at a 4 mg–20 mg/day dose was used for 3 to 5 days, instead of the MPD. Another 32 cases (24.7%) were treated only with high doses of oral prednisone (1–2 mg/kg/day). In one case, IV immunoglobulin was administered, because of lack of response to corticosteroid therapy.
11 In 10 cases (7.7%), there was spontaneous remission without treatment, and, in a further 10 cases (7.7%), there was no information on doses or routes of administration of the corticosteroids. Only 54 patients (41.8%) were tested for antithyroid antibodies after treatment. Of these, 46 (85.2%) presented normal levels after treatment, and, in 8 cases (14.8%), these levels remained high, even after completion of corticosteroid therapy. In 114 patients (87.6%), there was complete remission of symptoms after treatment; 8 patients (6.2%) did not show any improvement; 4 (3.1%) had partial improvement; and, in 4 cases (3.1%), there was no information on outcome after steroid therapy.
The patients' follow-up data were reported in 82 cases, with a duration ranging from 6 months to 2 years, and duration of follow-up not reported in 48 cases. In 48 cases (58.5%), there was no recurrence of disease during follow-up, and, in 22 cases (26.8%), relapse was associated with the withdrawal of corticosteroid; 12 patients (14.7%) relapsed, requiring further treatment, and 4 patients (4.8%) died.
DISCUSSION
Our data show that HE has a highly variable clinical spectrum, which may reflect difficulties in its recognition and early treatment and its poor outcome. Although there is a wide variation in age at the onset of clinical symptoms, affecting children, adults, and elderly patients, we observe a higher occurrence of the disease between the fifth and sixth decades of life. The progressive form seems to be the most common one, which further delays the diagnosis. Cognitive deficit and generalized seizures were the most common manifestations in diagnosis; this disease should be considered as an important differential diagnosis for acute and chronic progressive dementia and encephalopathy of unknown etiology.
The etiology of he remains unknown. Pathogenetic mechanisms include autoimmune cerebral vasculitis,
6,7 toxic effects of thyroid-stimulating hormone on the CNS,
1 and neuronal reaction mediated by antibodies.
8–10 autoimmune mechanisms are highly likely, owing to the higher prevalence in women, fluctuating course, and good response to steroids; however, it is still not known whether antibodies play a direct role in brain dysfunction or are simply markers of the disease. The vast majority of patients presented positive serum anti-TPO antibodies (86%), whereas anti-Tg was positive in fewer cases (48%). Blanchin et al.
8 have suggested that anti-TPO antibodies in the CSF and/or serum, and not anti-Tg, could contribute to the disease itself by interacting with CNS tissue, although we cannot rule out the pathogenic role of other antibodies. Our patient had high anti-TPO positivity in CSF and serum; however, despite what was observed in our review, there was no relationship between the levels and their severity or course of the disease after treatment. Most authors do not discuss the behavior of these antibodies in the CSF of their patients; however, among those who were tested, 75% (15 of 20) were positive for anti-TPO, and 85% (11 of 13) for anti-Tg in CSF.
Some researchers have attempted to demonstrate the role of common antigens against thyroid and brain tissues, such as antigen amino terminal [propto] enolase (NAE),
11 which proved to be highly prevalent (68%; 17 of 25) and specific in the population studied. This finding might help to explain the pathophysiology of the disease and also its diagnosis. Thyroid status does not appear to influence the mechanism of the disease, since most patients presented with normal thyroid function on diagnosis, as well as levels unrelated to the course of the disease. However, it is possible that a variation within the normal range could have an influence on the disease.
Unlike what its name might suggest, several cases of HE have been described in patients with Graves' disease. In our review of the literature, 19% of patients had this condition, which reinforces the fact that this disease is not associated exclusively with Hashimoto's thyroiditis, but also with other autoimmune thyroid diseases. For these reasons, it would be more appropriate to use the term “steroid-responsive encephalopathy associated with autoimmune thyroiditis (SREAT),” rather than Hashimoto's encephalopathy. The analysis of CSF may show a mild and nonspecific inflammatory state, both in patients with Graves' disease and in those with Hashimoto's thyroiditis who develop SREAT, in most cases, with normal or increased cellularity at the expense of lymphocytes.
5 the most characteristic findings, albeit not very specific ones, were the presence of anti-Tg and anti-TPO antithyroid antibodies and a high CSF protein concentration. Thus, in patients with encephalopathy of unknown etiology, these measurements in the CSF should reinforce the diagnosis. Nonetheless, we have to consider that these antibodies may be detected in low titers in 3% to 4% of the normal population.
1The EEG adds little to the diagnosis of HE, as none of the findings are specific. The most frequent changes found were a widespread slowing of waves and FIRDA. Slow focal waves could still be found. Generally, the degree of slowness of the waves is related to the clinical severity of the encephalopathy.
12 it should be considered that the normalization of EEG changes after corticosteroid therapy may allow the differentiation of uncertain cases from other diseases, such as Creutzfeld-Jakob and MELAS (mitochondrial encephalomyopathy syndrome, lactic acidosis, and stroke-like episodes).
13,14 Brain imaging shows no specific findings, but may contribute to the diagnosis of HE when it excludes other diagnostic possibilities.
5 The CT scan may be the first examination to be conducted. The changes are nonspecific, and they range from cerebral atrophy (the most common alteration) to ischemic abnormalities and/or cerebral edema. In more than 50% of cases, the MRI showed no changes. When it did, the most common finding was hyper-signal on T2, which may represent an edema or brain inflammation, and may be reversible after treatment with corticosteroids. This abnormality is nonspecific and is found in other causes of encephalopathy. In selected cases, methods that access the cerebral vascular function, such as magnetic resonance angiography and SPECT, can be used,
5 but the results so far have contributed little to the diagnosis. In view of this, it may be suggested that the diagnosis of HE should be based on neuropsychiatric manifestations associated with the following findings, provided that other causes of encephalopathy are excluded: 1) presence of high titers of antithyroid antibodies in the serum or CSF; 2) no change in CSF indicative of an infectious, vascular, or neoplastic etiology; 3) nonspecific MRI or CT scans; and 4) a good response to immunosuppressive therapy. We have observed that, in general, HE responds promptly to treatment and appears to be a disease with a good prognosis. There was complete remission of symptoms in most patients, followed by normalization of neuroimaging and EEG after corticosteroid therapy. However, there may be a form of partial remission, with persistence of the neurological and/or imaging changes, and even death from complications of the disease.
15 Treatment should be based on the use of corticosteroids, most commonly, IV methylprednisolone (MPD; 500–1,000 g/day) for 3 to 5 days, with oral prednisone at a high initial dose (1–2 mg/kg/day), followed by gradual reduction, based on clinical improvement. Other immunosuppressive drugs, such as azathioprine, may be used in association with steroids. This was the treatment chosen in our patient. Approximately 8% of the patients studied showed complete remission of symptoms, laboratory, and radiographic changes, and were stable without relapse during the entire follow-up, but there was no report of any clinical or laboratory characteristics that might identify patients likely to achieve this outcome.
8,16–18 Relapses can occur even after a significant improvement in symptoms or during the reduction in corticosteroid therapy. However, a return to the previous dose and prolonged maintenance have proved sufficient to control the signs and symptoms.
4,5,9,14,19–24 In some cases, the relapse occurred during follow-up after an asymptomatic period, making it necessary to adopt other measures in an attempt to control the disease, such as a course of plasmapheresis,
25–28 IV immunoglobulin,
11,27 methotrexate in doses of up to 15 mg per week,
29 cyclophosphamide pulses,
29 and even thyroidectomy.
30,31CONCLUSION
Although HE is a rare clinical condition, with a varying clinical course, it should be considered in any patient presenting acute or subacute encephalopathy of unknown etiology or in patients with diffuse cognitive decline with an intermittent or progressive course. Suspected patients, with or without active thyroid disease, after excluding the main causes, should be tested for antithyroid antibodies in serum or CSF.
Acknowledgments
The study presents no conflict of interest or funding.