Major depression remains a common disorder in late life (prevalence of 3%) associated with substantial morbidity and increased mortality. Cognitive impairments, especially in attention, learning and memory, and executive function, are frequent
(1–
3) and associated with poor outcome
(3,
4), and depression is a risk factor for subsequent dementia
(5). Impairments frequently persist, despite recovery from depression
(1,
2). The causes and associations of impaired cognitive function during depression remain uncertain. An influential hypothesis suggests that hypercortisolemia during depression may be important
(6). Studies in animals and humans, involving both exogenous steroid administration and conditions such as Cushing’s syndrome and stress, which raise endogenous levels
(7–
9), have shown a relationship between raised cortisol levels and neuropsychological dysfunction, especially memory impairment. Both glucocorticoid and mineralocorticoid steroid receptors are present in high concentrations in the hippocampus and frontal cortex, and prolonged and raised cortisol levels can produce neuronal dysfunction with decreased glucose uptake, reduced dendritic arborization, and, ultimately, neuronal death and cell loss in the hippocampus in animals
(6,
10,
11). It has, therefore, been proposed that raised cortisol levels during depression might be associated with cognitive impairments, especially in functions subserved by medial temporal lobe structures, and that persisting hypercortisolemia might cause hippocampal damage, explaining why impairments persist in depressed subjects, even when affective symptoms have resolved
(6,
11). Older subjects might be particularly susceptible to this process, since hypercortisolemia in depression is more common with advancing age
(12) and the aged hippocampus appears especially vulnerable
(13).
However, currently there is only limited direct evidence to support this hypothesis. A relationship between hypercortisolemia and impaired cognition has been described in some
(14), although by no means all
(15), studies. Hippocampal atrophy, sometimes unilateral, has been described
(16–
21), but other reports have found no evidence of atrophy in younger
(22) or older
(23,
24) subjects with depression. In a study of middle-aged depressed women, hippocampal volume reduction was related to total lifetime duration of depression
(18), a finding compatible with cortisol-induced neurotoxicity, although cortisol levels were not measured in that study. Others have found that hippocampal atrophy is present only after multiple depressive episodes
(25), although changes in first-episode patients may occur
(20). Genotype may be important, since hippocampal changes in depression have been linked to the apolipoprotein E-4 (APOE4) genotype
(26). Few previous investigators have examined hypothalamic-pituitary-adrenal (HPA) axis function in relation to these hippocampal changes, although there may be an age-related relationship between ventricular enlargement and hypercortisolemia
(27). Axelson and colleagues
(28) reported a relationship between lower hippocampal volume and higher cortisol levels with the dexamethasone suppression test, although overall they found no reduction in hippocampal volume in their depressed subjects.
Because of these inconsistencies in the literature, we performed a longitudinal study to test the hypothesis that hippocampal volume reduction occurred in older subjects with depression, with volume reduction that would be related to hypercortisolemia during depression and persisting cognitive impairments at follow-up.
Method
Subjects
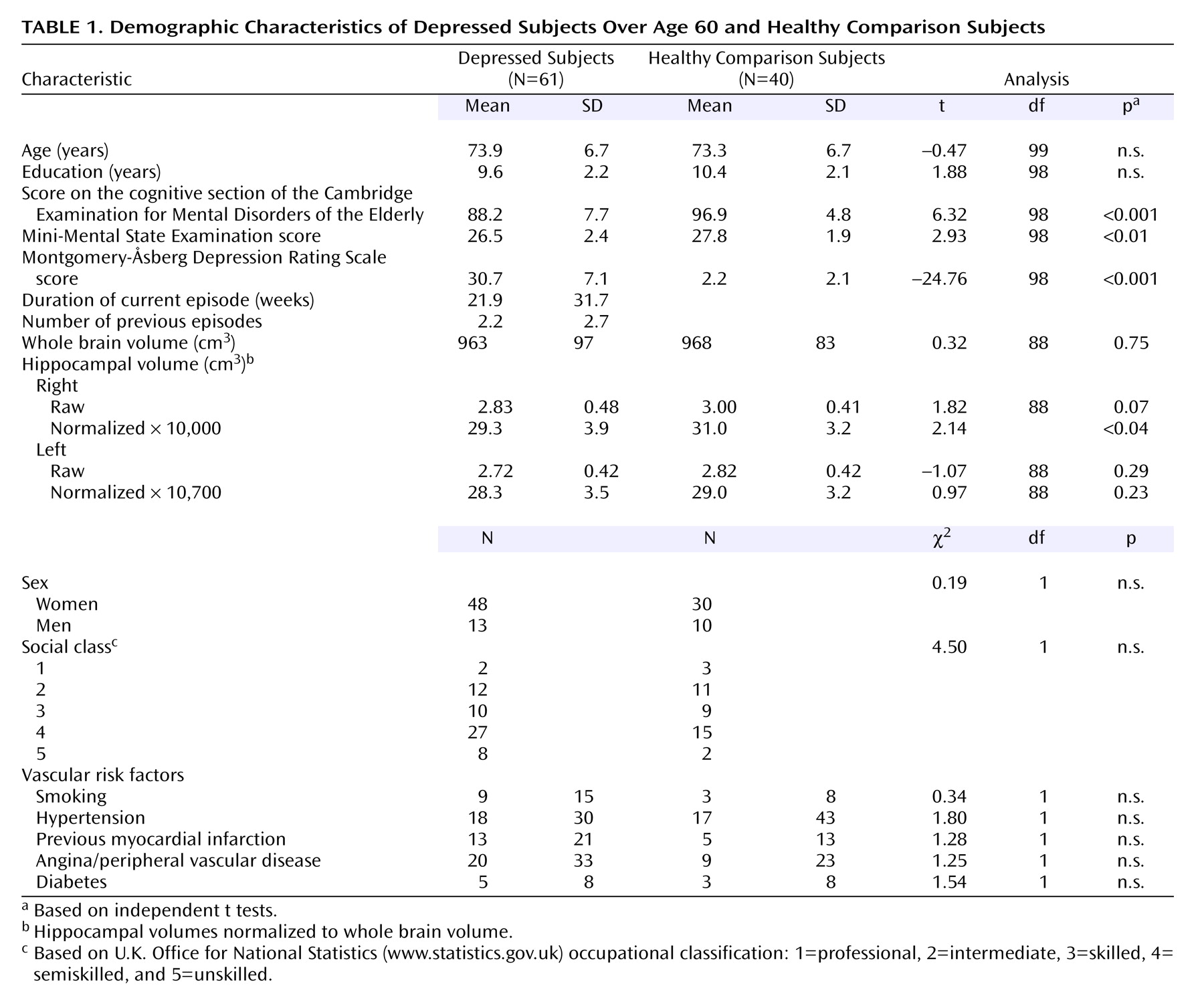
Sixty-one subjects ages 60 and over who fulfilled DSM-IV criteria for major depression (and scored 20 or more on the Montgomery-Åsberg Depression Rating Scale
[29]) were recruited from clinical old-age general psychiatry services covering geographically based catchment areas and included referrals from day hospitals, inpatient units, and outpatient clinics. A comparison group (N=40) of similar-aged older people (also all over 60 years of age) with no past history of depression or current depression (Montgomery-Åsberg Depression Rating Scale score of less than 8) were recruited from community sources such as the Royal British Legion and from the spouses of patients attending the same hospital units. Both subjects and comparison subjects with a history of prior cognitive impairment, history or evidence of stroke or transient ischemic attack, severe or unstable physical illness (e.g., insulin-dependent diabetes mellitus, untreated hypothyroidism, uncontrolled heart failure, cancer), or a cognitive section of the Cambridge Examination for Mental Disorders of the Elderly
(30) score of <75 (<80 for comparison subjects) were excluded from the study. The cognitive section of the Cambridge Examination for Mental Disorders of the Elderly is an extended cognitive screen (maximum score=107) that includes all of the items within the 30-point Mini-Mental State Examination (MMSE). Other exclusion criteria were history or current evidence of substance/alcohol abuse; long-term use (>2 months) of steroids at any point during lifetime; any use within the last 3 months of steroid or other medication thought to interfere with the HPA axis; ECT in last 3 months; use of medication that might significantly affect cognition (for example, use of benzodiazepines except short-acting ones, such as hypnotics, antipsychotics, sedative tricyclic antidepressants, or anticholinergic medication); or the presence of other neurological diagnosis. Use of newer antidepressants (e.g., selective serotonin reuptake inhibitors [SSRIs] and venlafaxine) and lithium was permitted. Fifty-one subjects were taking antidepressants at the time of testing, seven were taking lithium, and 19 had a past history of treatment with ECT (although they had had none in the last 3 months). The study was approved by the local ethics committee, and all patients and comparison subjects gave written informed consent to participate.
Assessment
Depressed subjects underwent a comprehensive psychiatric assessment, including a history, a mental status workup, a cognitive screen (cognitive section of the Cambridge Examination for Mental Disorders of the Elderly), and a physical examination. Subsequent investigations involved screening blood tests, including thyroid function, B12, and folate levels. Comprehensive demographic information was collected from all subjects and included past and current medical and psychiatric histories, including detailed analysis of medication taken, family history, education, and social class. For each episode of depression, case notes were searched and/or general practitioner records accessed, along with detailed patient and informant accounts to determine the number of previous episodes, age at onset, and the total lifetime duration of depression. Rating scales administered included the Montgomery-Åsberg Depression Rating Scale and the cognitive section of the Cambridge Examination for Mental Disorders of the Elderly and a neuropsychological battery to be detailed. A 10-ml venous blood sample was taken for genotyping.
Neuropsychological Assessment
The test battery was primarily designed to test attention, memory, and executive function and included both traditional pen-and-paper and computerized tasks
(31,
32). Tests administered were as follows:
1.
A computerized continuous performance task (VIGIL)
(33). Errors of commission and omission and response latency were recorded. This is primarily an attentional task.
2.
The FAS verbal fluency test, a task sensitive to frontal lobe impairment.
3.
Difference in performance between the Trail Making A and B tasks, a measure of executive function.
4.
Forward and reverse digit span. This task assesses attention and working memory.
5.
Rey Auditory Verbal Learning Test. This is a test of immediate and delayed verbal memory and of learning.
6.
Rey Visual Design Learning Test. This is a test of nonverbal memory.
7.
Memory tests from the Cambridge Automated Neuropsychological Test Battery (CeNeS Ltd., Cambridge, U.K.)
(32). This is a nonverbal battery (using abstract designs) that is automated in delivery, operates by using a touch-sensitive screen, and has been used in a number of previous studies of neuropsychiatric disorders
(34–
36), including in younger
(37) and older
(1,
2) subjects with depression. In the pattern recognition task, the subject has to recognize a series of 24 patterns; in spatial recognition, the subject has to recognize the position of a series of 20 squares; and in delayed matching to sample, the subject has to pick out a previously shown shape from three distracters after a delay ranging from 0 to 12 seconds.
8.
Cambridge Automated Neuropsychological Test Battery spatial memory span task.
9.
The Cambridge Automated Neuropsychological Test Battery spatial working memory test in which subjects search a number of boxes on a screen for hidden tokens. This task tests spatial working memory and also the need for a search strategy and thus central executive function.
10.
The Cambridge Automated Neuropsychological Test Battery Tower of London (Stockings of Cambridge) task. Subjects are presented with three “pockets” that can hold one, two, and three colored balls, respectively, and have to match one set with another by moving balls one at a time. This task has an attentional component but focuses on information manipulation and planning; thus it tests executive function and has been shown to activate the dorsolateral prefrontal cortex.
Salivary Cortisol Analysis
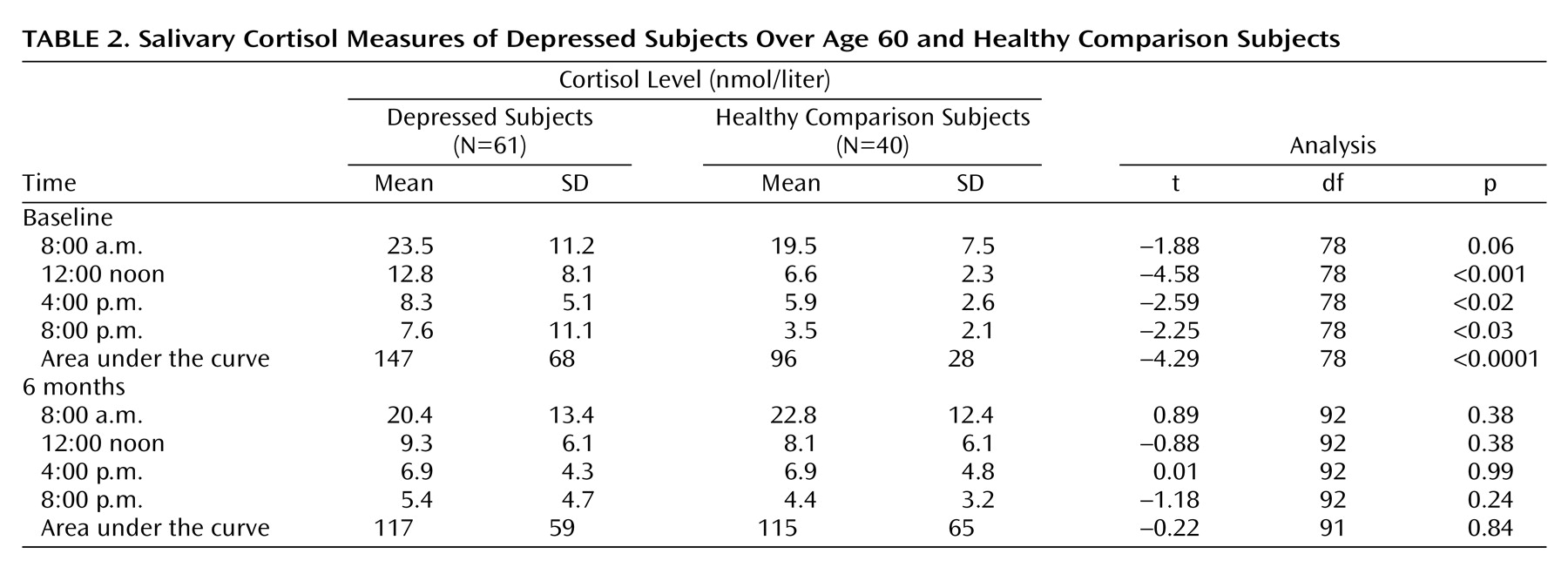
Assessment of cortisol levels was undertaken by using salivary samples collected by using Salivettes (Sarstedt, Nümbrecht, Germany). A Salivette consists of a plastic tube with a wool plug on which the subject chews to produce saliva. Samples were collected at four time points (8:00 a.m., 12:00 noon, 4:00 p.m., 8:00 p.m.) over 3 consecutive days in an attempt to obtain a comprehensive measure of cortisol production. For inpatients, samples were not taken within 1 week of admission, and for day- and outpatients, samples were taken in the subjects’ natural environment (i.e., at home). The Salivette tubes were centrifuged and the samples stored at –20°C until assayed. Salivary cortisol was measured by using an I125 disequilibrium assay, the radioactive cortisol for which was supplied by Amersham Health, Amersham, U.K., and the primary antibody Ab1002 and the solid phase antirabbit serum by IDS, Tyne and Wear, U.K. The intra- and interassay coefficients of variation for 7.0, 47, and 87 nmol/liter cortisol samples were 13.0% and 14.6%, 10.7% and 9.8%, and 9.4% and 10.2%, respectively. Average area under the curve for the 3 days was calculated.
Genotyping
APOE genotypes were analyzed by using polymerase chain reaction
(38).
Magnetic Resonance Imaging (MRI) Scanning Protocol
MRI scans were undertaken by using a 1.0 Tesla Siemens Magnetom Impact Expert System (Siemens Medical, Erlangen, Germany). Whole brain T1-weighted three-dimensional MPRAGE (magnetization prepared rapid-acquisition gradient echo) turbo flash data sets were acquired in the sagittal plane (TR=11.4 msec, TE=4.4 msec, TI=400 msec, flip angle=15°, matrix=256×256, slice thickness=1 mm, cubic voxels of 1 mm). The images acquired thus consisted of truly isotropic voxels of 1×1×1 mm. The sequences were chosen to give good gray-white matter contrast.
MRI Analysis
Images were transferred to a Sun Ultra 10 work station (Sun Microsystems, Mountain View, Calif.) running Solaris 2.7 and analyzed by a single operator (who was blind to diagnosis) using the commercially available software package AnalyzeAVW-3.0 (AnalyzeDirect.com, Lenexa, Kan.; Mayo Foundation Biomedical Imaging Resource, Rochester, Minn.). Images were reoriented along the long axis of the hippocampus to ensure consistent slicing of the hippocampus normal to its long axis in coronal sections. Hippocampal volume was assessed by using a region-of-interest approach with manual segmentation by use of a mouse. All identification of anatomical structures was carried out with reference to standard neuroanatomical and neuroradiological atlases and specific anatomical reviews
(39,
40). Hippocampal boundaries were based on previous descriptions
(41). The anterior limit was taken as the first slice in which the head of the hippocampus was visible. The alveus was used to aid demarcation of hippocampal gray matter from that of the amygdala (which is superior and anterior to it). The posterior limit was the slice on which the fornix was visible in its longest length. The superior boundary was defined by the choroid fissure, the medial boundary by CSF; the lateral boundary was the parahippocampal gyrus (for the head), the temporal horn of the lateral ventricle (for the body), the fornix (for the tail), and the inferior boundary by the subiculum (which was included in the measurement). A full protocol is available upon request from the first author. Whole brain volume was calculated by using the ANALYSE method, which uses a seed-growing thresholding approach with dilations and erosions. Final rendered brain volumes were compared with original scans for each subject to check accuracy. To assess intrarater reliability, repeat measurements were performed on 10 randomly selected scans on two occasions. Reliability for the volume of the segmented whole brain was excellent, with an intraclass correlation coefficient of α=0.99, and was very good for hippocampal volume (left hippocampus, α=0.97; right hippocampus, α=0.99).
Follow-Up
Depressed subjects and comparison subjects were reassessed at 6 months, with a repeat psychiatric assessment, administration of ratings scales, a neuropsychological assessment, and salivary cortisol tests.
For the purposes of analysis, depressed patients were considered fully recovered if they no longer met DSM-IV criteria for major depression and their Montgomery-Åsberg Depression Rating Scale scores were <8.
Statistical Analysis
Data were analyzed with SPSS version 10 (SPSS, Chicago). Differences between groups were assessed by using independent t tests or analysis of covariance when covariates (e.g., age) were included. Paired t tests were used to investigate differences over time within groups. To deal with the potential problem of multiple comparisons (particularly when investigating correlations between cognitive test results), hippocampal volume, cortisol level, and cognitive tests involving memory were combined into a single z score. For the five tests chosen to reflect memory/hippocampal function (the Rey Auditory Verbal Learning Test, the Rey Visual Design Learning Test, pattern recognition, spatial recognition, delayed matching to sample), the mean for the comparison subjects at that particular time point (baseline or the 6-month test) was subtracted from the raw score for each subject, and the result was divided by the comparison’s standard deviation. This created z scores for the comparison subjects (mean=0, SD=1) for each of the five tasks; performance of the depressed patients was measured relative to this. The five memory z scores were then summed to create an overall memory z score (zMem), which was used as the primary variable for correlational analysis. zMem scores were calculated for memory at both baseline and 6 months. Bivariate correlations were undertaken using Pearson’s r, with partial correlations in some cases for age adjustment. Linear regression was used to investigate the predictors of continued cognitive impairment at 6 months in depressed subjects. The significance level was set at p<0.05.
Discussion
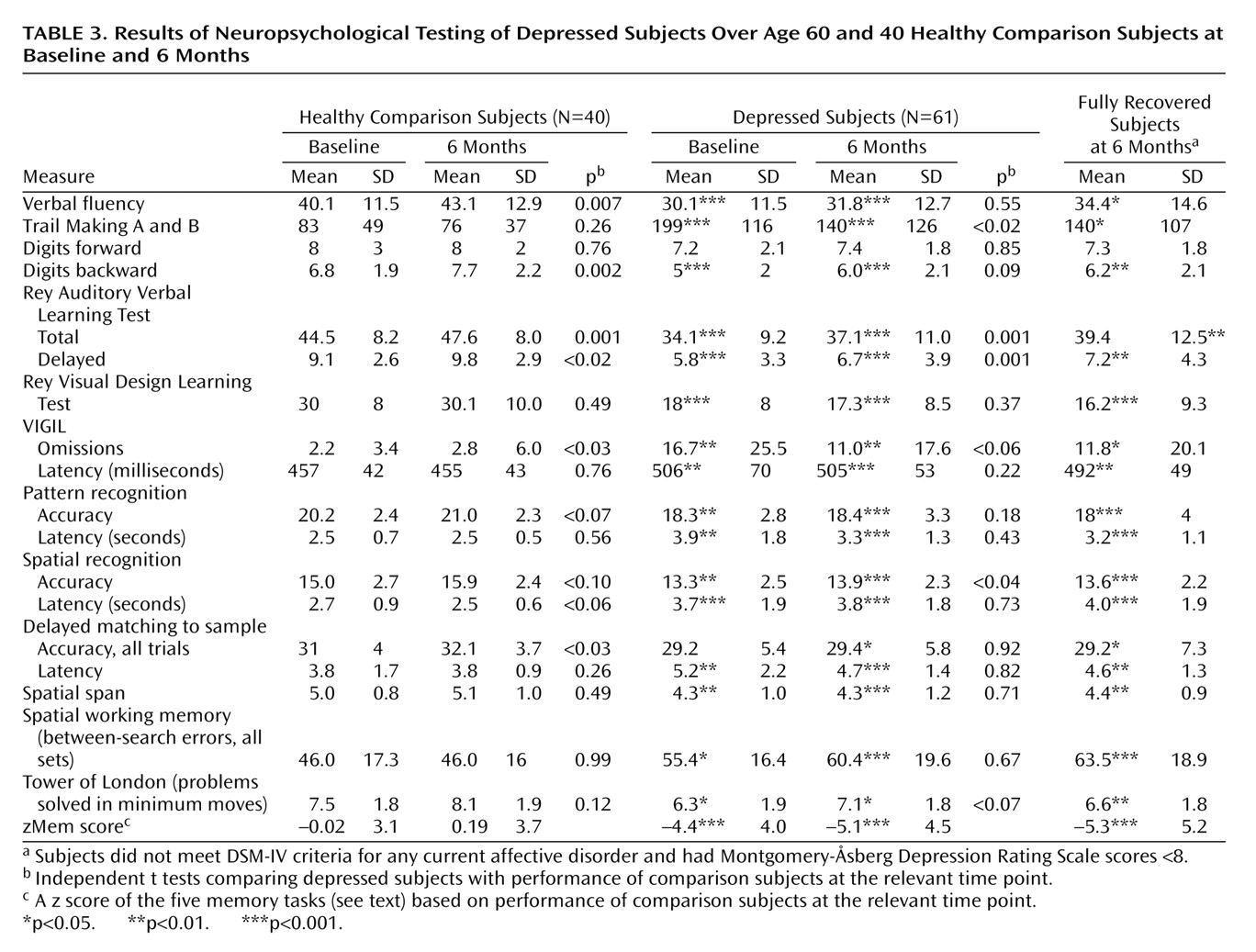
The main results of this study were that 1) in relation to similar-aged comparison subjects, older depressed subjects had multiple cognitive deficits in attention, learning and memory, and executive function; 2) deficits persisted at 6 months, even in subjects who were in full remission from illness, with 41% fulfilling criteria for mild cognitive impairment; 3) depressed subjects exhibited hypercortisolemia during depression, as demonstrated by raised salivary cortisol levels, with values returning to normal by 6 months; 4) depressed subjects had significant volume reduction of the right hippocampus on MRI; 5) continuing deficits in memory were associated with bilateral hippocampal volume reduction, not cortisol levels, APOE genotype, or current level of depressive symptoms.
Our first hypothesis—that depressed subjects would have reduction in hippocampal volume on MRI—was confirmed for the right hippocampus. Hippocampal volume reduction has been found in previous studies in younger and older depressed patients
(16,
19–
21,
25,
43,
44), although negative results have also been reported
(22,
45–47). Unilateral change has also been reported in many of these studies. Intriguingly, studies of younger subjects tend to find preferential involvement of the left hippocampus
(19,
44,
48), while studies of older subjects, in keeping with our results, find more involvement of the right hippocampus
(16,
21). Unilateral changes would tend not to support the glucocorticoid toxicity hypothesis, since there would be no good reason why a systemic toxic process would cause unilateral brain damage. Right hippocampal volume is larger than left and so potentially may be more susceptible to age-related atrophy, although a more likely explanation may be that changes are indeed bilateral but studies may lack the power to demonstrate bilateral changes. In support, most studies, including this one, that find unilateral changes find reductions, but to a lesser degree, on the other side. The finding of hippocampal atrophy in depression receives some support from a recent neuropathological study that, while not finding neuronal loss, did find increased cell packing, consistent with loss of neuronal processes
(49).
As expected, we found clear evidence of increased cortisol production (53%) in depressed subjects in relation to comparison subjects, with levels returning to comparison values by 6 months, paralleling the substantial improvement in depression. Despite reversal of hypercortisolemia and improvement in depression, significant impairments in multiple cognitive domains persisted at 6 months. The possibility that such impairments were the result of residual depressive symptoms was refuted by finding that results were essentially unchanged when only fully remitted subjects were examined (
Table 1). Previous studies have emphasized the broad nature of cognitive impairments in depression, and impairments on the same or similar tasks to those included here have been described by many others
(1–
3,
50–52). Although most of our subjects were taking medication, we do not think that medication effects accounted for these deficits for several reasons. First, prescriptions for tricyclic antidepressants and sedative benzodiazepines were exclusion criteria; in comparison, SSRIs have few adverse effects on cognition
(53). Second, the relationship between impairments and structural brain changes would not support drug-induced impairments. Third, a study of entirely drug-free depressed subjects reported an almost identical profile of neuropsychological impairment to that found here
(54).
Most authors find some cognitive improvement on recovery from depression, although impairments persist in many domains
(1,
2,
51,
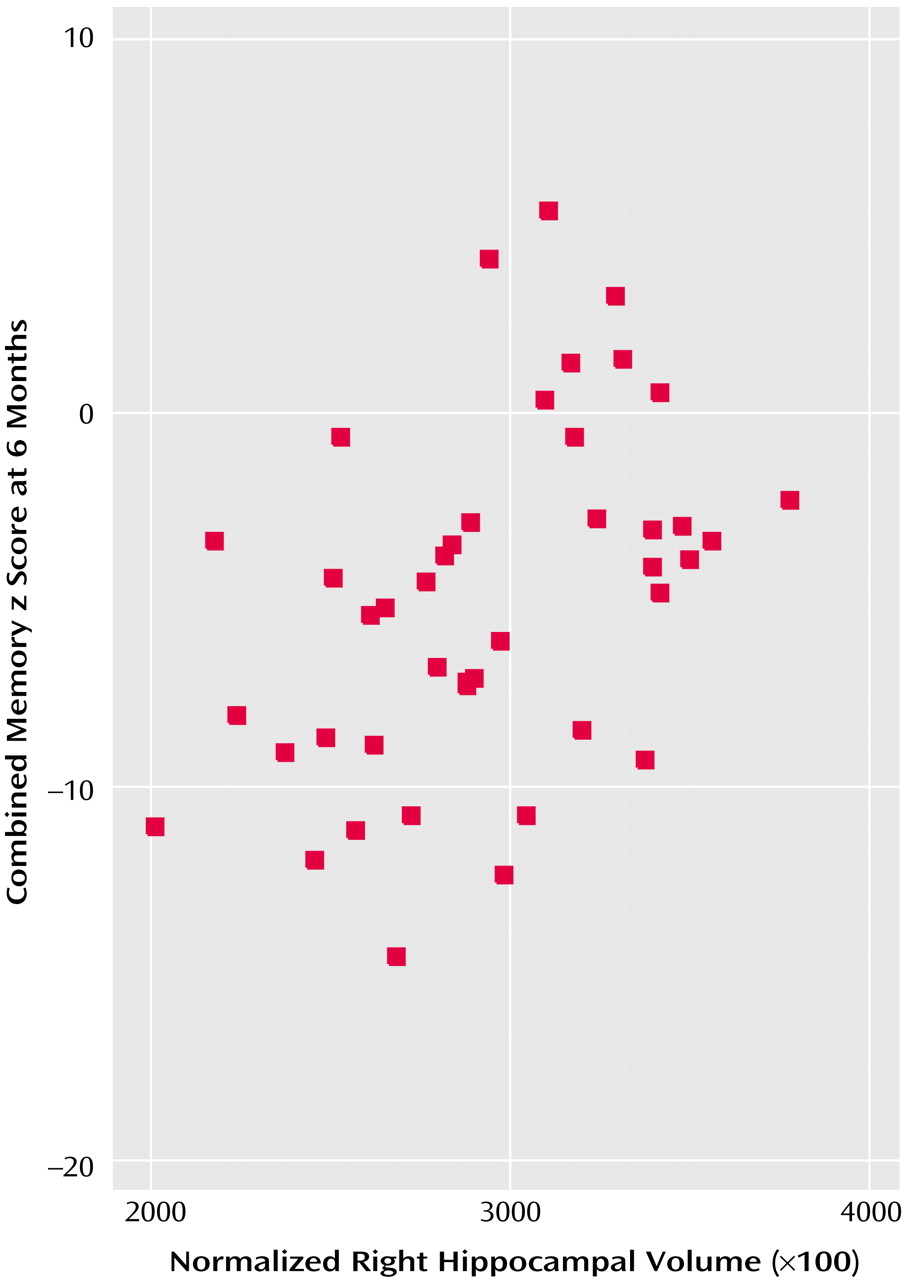
55). Few previous studies have retested comparison subjects to account for learning effects. The current investigation did, and we found surprising improvements in many tests of comparison subjects. These not only were in a greater number of tests than for depressed subjects but made the lack of improvement in cognition after improvement in depression all the more striking. In patients, deficits remained in attention, executive function, working memory, verbal and visual memory, new learning, and speed of response. Our main aim was to investigate the correlates of this continued impairment, with the hypothesis that impairments in learning and memory would be associated with hypercortisolemia during depression and with hippocampal volume reduction. We did find a significant association between hippocampal volume and persisting impairments, whether examined as a continuous measure (
Figure 1) or by dividing subjects into those with and without mild cognitive impairment. In both cases, the relationship remained after adjustment for age. Hippocampal volume reduction has also been described in nondepressed subjects with mild cognitive impairment and may predict cognitive decline
(41,
56). However, psychiatric illnesses, such as depression, have to date excluded subjects from the label of mild cognitive impairment
(42). We found that 41% of depressed subjects at 6 months fulfilled criteria for mild cognitive impairment, which appeared to be associated with hippocampal volume reduction, not residual depressive symptoms. It will be important to undertake longer-term follow-up of depressed patients with mild cognitive impairment described here to determine whether persisting impairments and/or hippocampal volume reduction predicts subsequent dementia. In a prospective study, Steffens and colleagues
(57) found that left hippocampal volume reduction was associated with subsequent cognitive decline in older subjects with depression. Further similar studies will enable outcome of depressed patients with impairments to be compared with those with amnestic impairments in the absence of depression, who are known to be at a higher risk of cognitive decline and dementia
(42).
Contrary to our second hypothesis, we did not find evidence of a relationship between raised cortisol levels during depression (or at recovery) and cognitive performance or hippocampal volume reduction. Our results, therefore, did not provide support for the glucocorticoid toxicity hypothesis of hippocampal damage in depression. Despite the studies cited describing hippocampal volume reduction in depression, there has not been a clear demonstration that this relationship is mediated by glucocorticoids. Moreover, cognitive deficits in depression have been well described, even in first-episode patients
(18,
25) who might be expected to have had relatively low exposure to raised glucocorticoids compared with those with multiple episodes. Hippocampal volume reductions have been described in many psychiatric disorders, including dementia, schizophrenia
(58), and posttraumatic stress disorder
(59). It is unlikely that glucocorticoid toxicity underlies such changes in all disorders. In addition, in other studies, hippocampal atrophy was found to be associated with late age at onset and APOE4 genotype
(16,
26,
57), suggesting that mechanisms other than raised cortisol levels might be important. Although we found no relation with APOE4 genotype, the absence of a relationship between hippocampal volume and either cortisol levels, number of previous depressive episodes, or lifetime duration of depression argues against cortisol toxicity.
There are a number of possible reasons for our negative findings. The limitations of our study include a relatively modest-sized and heterogeneous study group, as any subtype-specific correlations (for example, with psychotic or melancholic type) might have been diluted by the inclusion of other subjects. Our cortisol assessment, although rigorous, may not have accurately reflected total glucocorticoid exposure over a lifetime, which may be the real variable of importance. Individuals may vary in their susceptibility to raised cortisol levels, with such differences masked by the use of mean values for analysis. There may be other steroids that are important but were not assessed in this study, for example, dehydroepiandrosterone (DHEA). Age-related decrements in the hippocampal glucocorticoid receptors occur, and it may be that such changes are adaptive, actually protecting the aging hippocampus against glucocorticoid toxicity
(13). Finally, it may be that older subjects with depression differ from those at other ages. For example, the role of vascular factors in the etiology of late-onset depression has recently been recognized
(60,
61), while the increasing importance of vascular factors and cerebrovascular disease in underpinning cognitive impairments in a variety of disorders is recognized within the term “vascular cognitive impairment”
(62). It may be that vascular factors, rather than raised cortisol levels, may underpin structural brain changes and enduring cognitive impairments. However, in the current study, we found no differences between groups in the presence of established vascular risk factors, such as diabetes, smoking, and hypertension, although it remains possible that, as demonstrated in autopsy studies, depressed subjects might still have had more vascular disease than comparison subjects
(63).
In conclusion, we found evidence in a substantial number of older depressed subjects of persisting cognitive impairments that were linked to hippocampal volume reduction rather than current or past depressive episodes or hypercortisolemia. Longer-term follow-up is required to determine if such changes do indeed robustly predict subsequent cognitive decline, while neuropathological studies are required to determine the pathological substrates underlying hippocampal atrophy and persisting cognitive impairments in older depressed subjects.