Clozapine shows a unique profile of treatment efficacy, but cross-sectional and naturalistic studies link clozapine treatment to hyperglycemia and a greater relative risk of diabetes
(1–
3). Clozapine treatment has been associated with higher insulin levels than typical antipsychotics, suggesting that clozapine induces insulin resistance
(2). To our knowledge, glucose-insulin homeostasis has not been previously evaluated before and following clozapine treatment. Therefore, we tested the following hypotheses in a prospective study: 1) Clozapine treatment is associated with the development of abnormal glucose tolerance (as measured by an oral glucose tolerance test [GTT]). 2) Clozapine treatment is associated with the development of insulin resistance (measured by homeostasis model assessment).
Method
The South London and Maudsley National Health Service Trust research ethics committee approved the study. All patients within the hospitals’ catchment area intending to start taking clozapine during the years 2000–2001 were invited to participate. After complete description of the study to the subjects, written informed consent was obtained.
Inclusion criteria were DSM-IV diagnosis of schizophrenia and switching to treatment with clozapine as the sole antipsychotic medication. Exclusion criteria were diabetes mellitus, treatment for glycemic control, and conditions associated with glucose intolerance.
Twenty-eight patients had baseline assessments and began to take clozapine. Four dropped out because of hypotensive reactions, and two declined to participate further. The mean age of the remaining 22 subjects was 30.5 years (SD=7.4), 11 were female, 11 were male, 11 were black African or African/Caribbean, and 11 were white British. Sixteen patients smoked tobacco, and 17 drank alcohol (all drank less than 21 units/week).
Four patients had a family history of diabetes (in a first-degree relative, N=1; in a second-degree relative, N=3). Patients consumed a standard diet containing more than 150 g carbohydrate/day, and all but two were inpatients throughout the study.
The patients were given a baseline assessment in the week before they started to take clozapine. Clozapine dose was titrated according to clinical response. Patients were followed up after 2–4 months of treatment (mean=2.5 months, SD=0.95). At follow-up, the measures carried out at baseline were repeated, and clozapine levels were measured.
A standard oral GTT was performed according to World Health Organization (WHO) specifications
(4). Subjects fasted under observation from midnight before the test. Venous blood samples were taken at 9:00 a.m. to measure fasting plasma glucose, insulin, and clozapine levels. Subjects then drank 75 g of anhydrous glucose in 300 ml of water within 5 minutes, and venous blood samples were taken after 30 minutes (confirming glucose absorption) and after 2 hours. Plasma glucose was measured with an enzymatic reference assay (Cobas Integra 400, Roche Diagnostics, Lewes, East Sussex, U.K.) and classified according to WHO criteria
(4). Insulin was assayed with an ELISA kit (Mercodia Iso Insulin ELISA, Diagenics, Bletchley, Milton Keynes, U.K.). Body mass index (weight [kg]/height
2 [m]), diet, and medications were recorded.
Insulin resistance was evaluated with the homeostasis model assessment method: (fasting insulin [μU/ml] × fasting glucose [mmol/liter])/22.5
(5). Clozapine levels were measured with high performance liquid chromatography.
Paired-sample t tests were used to compare mean fasting glucose, insulin resistance, and 2-hour glucose levels before and after clozapine treatment. The 2-hour glucose levels were adjusted by using simple linear regression (dependent variable: change in 2-hour glucose level; independent variable: change in fasting glucose level). The McNemar test for paired proportions was used to compare the ratio of abnormal to normal oral GTT results before and after clozapine treatment. Correlations between change in body mass index and change in fasting and 2-hour glucose levels were tested with Pearson’s product moment correlation.
Results
Two patients were diabetic at baseline and excluded. Of the remaining 20 patients, 11 (55%) developed de novo abnormal glucose tolerance on oral GTT measurement (one diabetes mellitus, eight impaired glucose tolerance, and two impaired fasting glycemia). There was a highly significant increase in the proportion of patients showing abnormal glucose control following clozapine compared with before treatment (p=0.006, McNemar test).
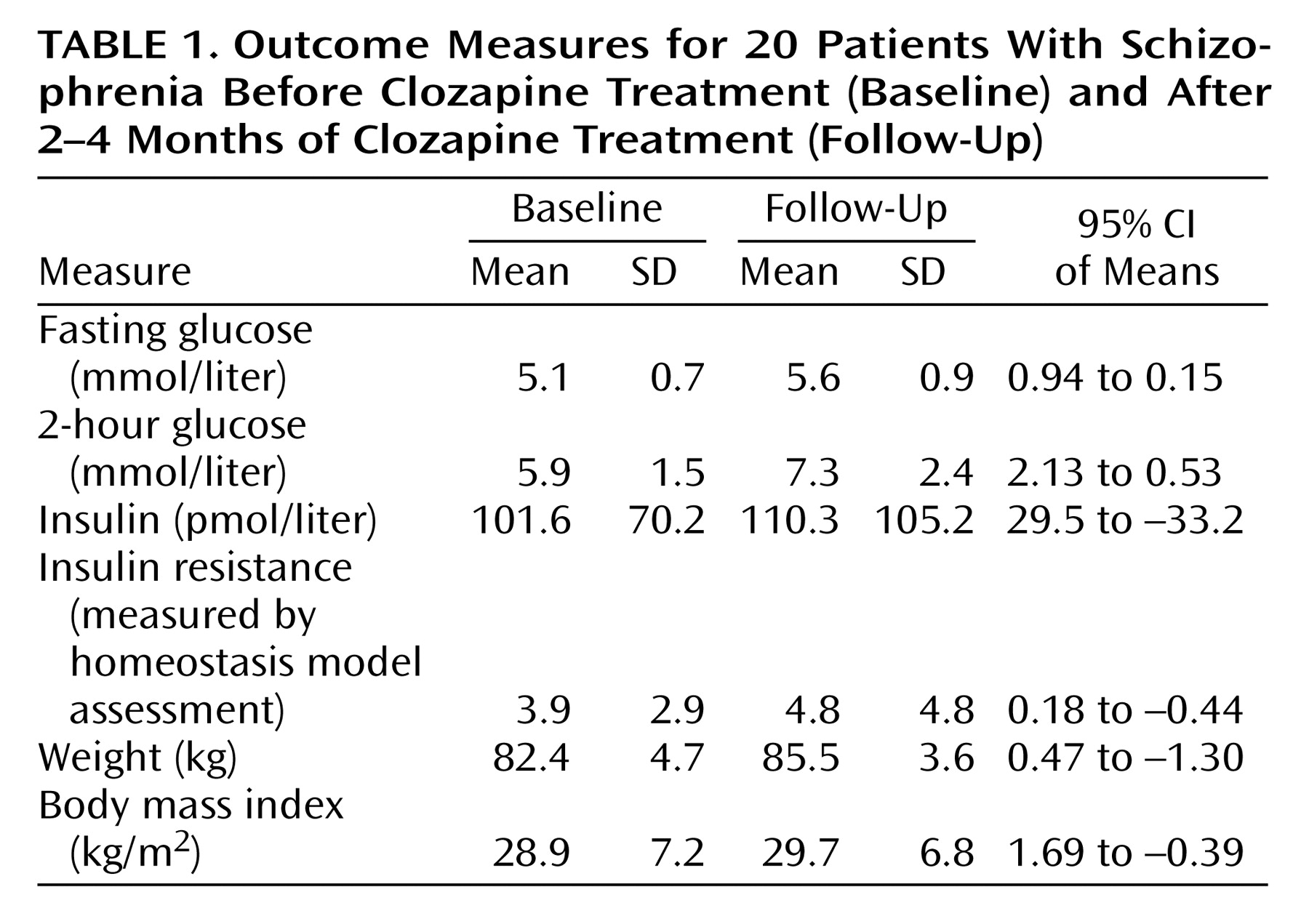
Table 1 shows baseline and follow-up levels for the outcome measures.
Mean fasting glucose level increased by 0.55 mmol/liter (t=–2.9, df=19, p=0.01), and mean 2-hour glucose level increased by 1.4 mmol/liter (t=–3.5, df=19, p=0.002); this difference remained significant after adjustment for fasting glucose (t=–3.2, df=19, p=0.005). There was no significant change in insulin level (t=0.128, df=14, p=0.9) or insulin resistance level measured with the homeostasis model assessment (t=–0.9, df=14, p=0.37). Mean body mass index increased by 0.82 kg/m2, although this was not significant (t=–1.325, df=17, p=0.2), and there was no correlation between change in body mass index and change in fasting (r=0.17, N=20, p=0.49) or 2-hour (r=0.37, N=20, p=0.14) glucose levels.
Medications at baseline were olanzapine (N=10), amisulpride (N=1), quetiapine (N=1), risperidone (N=3), zuclopenthixol (N=3), sulpiride (N=2), venlafaxine (N=4), paroxetine (N=1), fluoxetine (N=2), orphenadrine (N=2), procyclidine (N=2), carbamazepine (N=1), lithium (N=1), sodium valproate (N=1), beclomethasone inhaler (N=1), and omeprazole (N=1).
At follow-up the mean clozapine dose was 341 mg/day (SD=105.4), and the mean clozapine blood level was 0.42 mg/liter (SD=0.21). Apart from switching antipsychotic to clozapine, there were no changes in patients’ drug regimens from baseline.
Discussion
To our knowledge, this is the first direct evidence that, within 4 months, clozapine treatment results in increased plasma glucose concentrations, independent of changes in insulin resistance or body mass index. Clozapine treatment was associated with a change in glucose concentration of 0.65–0.85 standard deviations and the onset of abnormal glucose tolerance in 55% of patients as assessed by oral GTT. This study extends previous findings (1–3) by indicating that de novo glucose control abnormalities are common within months of clozapine initiation in a group screened to exclude preexisting diabetes. Thus, it provides evidence for a causal relationship between clozapine treatment and the development of hyperglycemia.
Baseline body mass index was in the overweight range (25–30 kg/m2), consistent with previous studies of patients beginning to take clozapine (1). The baseline insulin resistance level was within the upper quintile of the range of insulin resistance levels seen in healthy control subjects, indicating a greater risk of developing diabetes (5). The similarity of insulin resistance levels to those found in previous reports (3) suggests that patients show a preexisting insulin insensitivity that may be related to previous treatment, schizophrenia, or raised body mass index.
It is possible that the development of hyperglycemia is unrelated to clozapine treatment and incidental or secondary to concomitant medication. However, concomitant medications were unaltered in these patients, and the short time span and magnitude of the changes make it unlikely that we are observing the natural development of abnormal glucose control: less than 5% of patients taking olanzapine developed hyperglycemia over a follow-up period of more than 2 years (2).
Insulin resistance levels did not show significant increases, which refutes the initial hypothesis that changes in glucose control following clozapine treatment are attributable to increased insulin resistance. Body mass index did not significantly increase. We did not measure changes in body composition; however, these characteristically influence glucose homeostasis through altered insulin sensitivity (6). An alternative mechanism would be a direct drug effect on glucose regulation. Clozapine reduces glucose uptake in neuronal cells (7). This would reset the plasma glucose levels that glucose-sensing neurones accept as normal, meaning the glucose level is perceived as lower than it is and resulting in compensatory increases in glucose levels. This mechanism would explain the rapid increase in plasma glucose level independent of changes in insulin resistance levels.
Our results indicate that patients taking clozapine are at greater risk of developing abnormal glucose control. Diabetes mellitus is a leading cause of morbidity, and patients with mental disorders often do not receive appropriate diabetic care (1, 6). There have been cases of diabetic ketoacidosis and death following clozapine treatment (3). Furthermore, hyperglycemia below the threshold for diagnosing diabetes is associated with greater risk of cardiovascular complications (6). Clinicians will need to weigh these issues in the balance when deciding to initiate treatment with clozapine.