ADHD is one of the most common heritable mental health disorders of childhood. Molecular analyses have identified a number of genes that appear to be involved in the etiology of ADHD
(1), and genome-wide scans have identified multiple regions of interest
(2 –
5) . However, many of the identified risks are unreplicated or of small effect. For example, of the many studies of the dopamine receptor gene DRD4, almost half failed to replicate association or linkage, and observed effect sizes have been small
(6,
7) . Similarly, the regions of interest identified through recent genome-wide scans show little overlap across studies, and no findings are significant at a genome-wide level.
The Role of Endophenotypes
One factor hampering progress in the study of ADHD is sample heterogeneity arising from etiological and phenotypic complexity
(8,
9) . The use of endophenotypes to reduce heterogeneity or identify more specific gene-behavior linkages is one strategy for managing this complexity and improving the power of molecular analyses
(10,
11) . Endophenotypes, which are more proximal markers of gene action in the same biological pathway linking genes and complex clinical symptoms, are thought to be less genetically complex than the broader disease phenotype, indexing a limited aspect of genetic risk for the disorder as a whole
(10,
12 –
14) .
Genes affect complex behavior through their influence on the development and function of the essential building blocks of the nervous system, such as neurons, transmitter systems, and neural networks. Hence, heritable variation in mental functions may reflect variations in the code specifying the structure and function of the underlying neural network. The various cognitive deficits observed in ADHD are thought to represent one major manifestation of this principle
(15) and are thus prime candidates for an endophenotype approach.
While the ultimate validity of a candidate endophenotype lies in its ability to increase the power to detect functional genetic variants in a disorder, we argue that a valid endophenotype must meet several criteria before it is included in genetic studies
(12) . An endophenotype should be common in affected individuals (sensitive), relatively unique to the disorder (specific), and comparatively uncommon among unaffected individuals in the general population. It should be functionally related to the pathobiology of the disorder. The endophenotype should be present in the genetic relatives of affected individuals and covary among them, with the likelihood of expression proportional to the degree of genetic relatedness between them. This should be true regardless of the affection status of the relatives; independence between overt clinical symptoms and the endophenotype demonstrates the capacity to detect asymptomatic genetic carriers or those with incomplete penetrance of the contributory genetic factor
(14,
16) .
Endophenotypes can be used as trait markers for disease susceptibility, to identify more genetically homogeneous subgroups, to highlight distinct pathophysiological mechanisms or etiological pathways, or to define “spectrum” phenotypes suitable for quantitative trait analyses
(12,
13) . Twin models suggest that continuous traits provide substantially more power to detect genetic effects than do binary or ordinal traits
(17) . Overall, the net effect of this approach is an increase in statistical power to detect genetic linkage or association through the use of more sensitive and specific sample selection criteria and the emancipation of sample selection from clinical diagnosis
(8,
13) .
Behavioral Inhibition as a Cognitive Endophenotype in ADHD
Motor response inhibition is a crucial executive function that comes into play when one tries to withhold or interrupt an ongoing or planned response when required to do so by external circumstances or by changes in intention. Frequently studied in ADHD using the stop-signal task, motor response inhibition meets many of the established criteria for a valid endophenotype
(12) .
Deficient response inhibition measured in the stop-signal task is a replicated and fairly specific deficit in ADHD
(18 –
24) . Response inhibition has a well-established neurological basis that overlaps considerably with the proposed biology of ADHD
(25 –
27) . A deficit in motor response inhibition has been identified as a familial marker for ADHD risk
(28), and several studies have documented an inhibitory control deficit in the unaffected siblings of children with ADHD
(29,
30) . Inspection of sibling dyads shows cosegration of inhibitory control ability
(31) . Finally, molecular studies have found links between deficient inhibition and dopamine system genes
(32 –
36) . Taken together, these studies demonstrate that an inhibitory deficit is more predictive than many other features in identifying ADHD risk and confirm the validity of inhibitory control as an endophenotype. No other cognitive ability has been as extensively validated in an endophenotype model as motor response inhibition.
Extension of the Endophenotype Model
If inhibitory control is an index of shared genetic risk, assessments of an inhibition deficit in children with ADHD and their parents could support the validity of the measure, much as sibling studies have done to date. However, the relationship between parents and children can also provide unique insights regarding the transmission of genetic risks. Siblings share 50% of their genes on average. Depending on the transmission of genes from each parent, some sibling pairs will actually share no genes while others may share close to 100%. Parents and their children also share 50% of their genes, but this value does not represent an average—half of the genes in a child are represented in each parent with 100% certainty. Furthermore, parental contributions may be quite distinct for genes located on the sex chromosomes or subject to sex-based regulatory phenomena, such as genomic imprinting, wherein gene expression in offspring varies on the basis of parental origin (see reference
37 for a review). Covariation between parents and children in inhibitory ability has not yet been investigated.
Preliminary evidence suggests that imprinting may play a role in the etiology of ADHD. Hyperactivity in mice was the first recorded behavioral effect of imprinted genes
(38), and a number of human neurological disorders that commonly occur in conjunction with ADHD show variations in symptoms or severity based on the parent of origin, such as Tourette’s syndrome
(39) and bipolar affective disorder
(40) . Goos et al.
(41) found phenotypic differences among children with ADHD depending on the sex of the parent who had a history of the disorder, and data published by Nigg et al.
(15) showed substantial differences in the correlation of cognitive scores between ADHD probands and parents of each sex. Finally, a number of genes relevant to the functioning of neurotransmitter systems implicated in ADHD and/or inhibitory control have shown preferential transmission of paternal alleles to affected children, indicative of genomic imprinting
(42 –
46) .
In this study, we address a significant gap in the literature by comparing inhibitory control in children with ADHD, their siblings, and their parents. Based on the endophenotype model of shared genetic risk, we predicted a deficit in inhibitory control in the parents of children with ADHD, independent of ADHD symptoms. We also predicted that inhibitory control ability would covary between parents and children, with parental ability significantly predicting child ability, independent of symptom severity in either generation. Finally, we explored the parental origin of this shared genetic risk, predicting that children’s inhibitory control ability would be influenced more by paternal than maternal inhibitory ability, in keeping with evidence of greater paternal transmission of risk alleles in ADHD.
Method
Participants
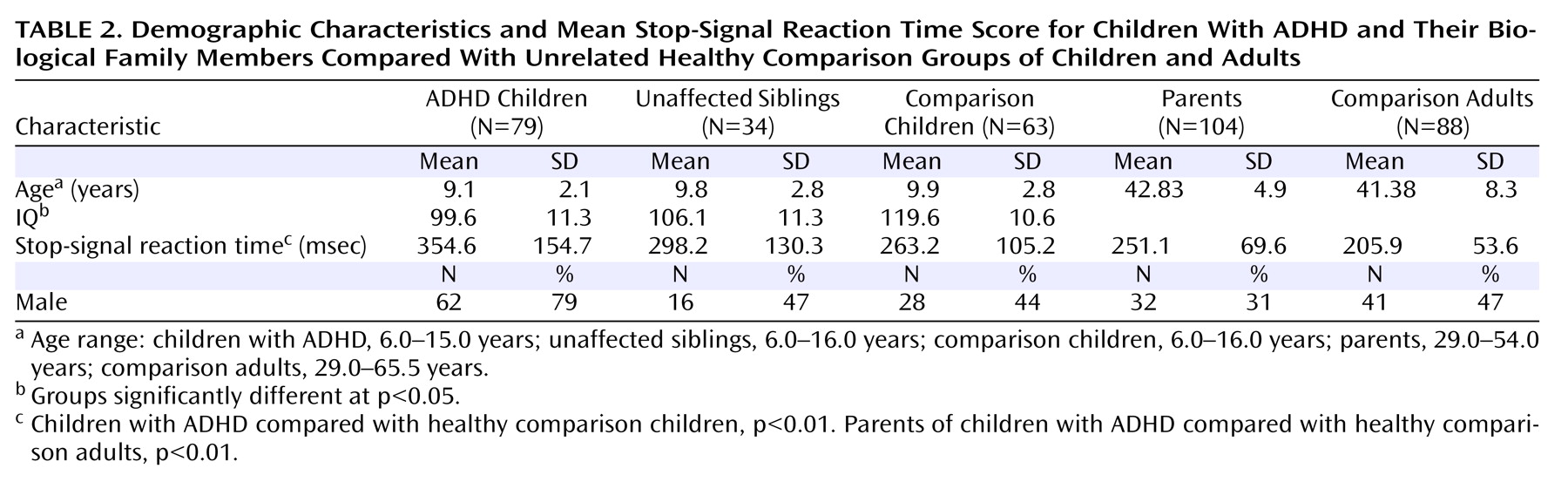
Seventy-five families who had been referred to a child psychiatry clinic in an urban pediatric hospital participated in the study. Each family included at least one child who met rigorous diagnostic criteria for ADHD. Forty families contributed one child to the sample; of these, 32 were affected and eight were unaffected siblings of an affected child whose data were excluded from the study because of inclusion and exclusion criteria (see below). Ten families contributed two affected children, 21 families contributed one affected and one unaffected child, one family contributed two unaffected siblings of an affected child who was excluded from the sample (as above), and three families contributed three children, at least one of whom was affected. In total, 113 children (69% male) ranging in age from 6 to 16 years (mean age=9.3 years [SD=2.3]) participated in the study; 79 of them (70%) met DSM-IV criteria for a diagnosis of ADHD. Healthy comparison children (N=63; mean age=9.97 years [SD=2.8]; 44.4% male) were recruited through local advertising. The referred children differed significantly from comparison children in mean IQ but not in mean age. IQ was included as a covariate in child group analyses.
Inclusion criteria were valid stop-signal reaction time scores for the child and for at least one parent. Children were excluded from the study if they had a history or evidence of neurological disorders (e.g., epilepsy), chronic or serious medical problems, psychosis, clinically significant mood or anxiety disorders, suspected genetic diseases, or IQ below 80 or above 130, or were taking medications that could not be withdrawn or have extended washout periods. The healthy comparison children met all of these criteria with the exception of an ADHD diagnosis and the participation of their parents. Although the majority of children seen in this clinic present for initial diagnosis and are not on any medications, the minimum medication withdrawal period in advance of participation in the study was 48 hours, which is sufficient for the dissipation of any effects on cognition
(47 –
50), and withdrawal periods were usually longer.
Participating families included 72 biological mothers (mean age=42.5 years [SD=4.6]) and 32 biological fathers (mean age=43.6 years [SD=5.6]); 29 families included both parents. Assessments of the healthy comparison children are part of an ongoing research control sample protocol and do not include parents. Therefore, for this initial effort, separate comparison groups of children and adults were obtained. A community sample of comparison adults (N=88; mean age=41.38 years [SD=8.32]; 46.6% male) was recruited at an educational installation at the Ontario Science Centre. Adult groups did not differ significantly in age.
The study sample was similar in demographic characteristics to the urban population from which it was drawn. Socioeconomic status was not assessed, although previous research has found this measure to be uncorrelated with measures of inhibitory control or related measures of cognitive task performance
(21) . The entire study sample was prospectively obtained and has not been presented previously.
Measures
Children underwent an extensive multistage screening and diagnostic process involving multiple independent and converging sources of information and a variety of assessment tools, including validated, age-appropriate, and standardized parent and teacher clinical interviews (the Parent Interview for Child Symptoms
[51] and the Teacher Telephone Interview
[52] ). Children who were classified as having ADHD met DSM-IV criteria for diagnosis, defined as having at least six of nine inattentive or six of nine hyperactive-impulsive symptoms, or both, with symptom onset before age 7. To ensure that moderate impairment was present in two settings, as required by DSM-IV criteria, children had to meet criteria for ADHD in the parent or the teacher interview, exhibit a minimum of four ADHD symptoms according to the second informant, and have at least a “moderately impaired” rating on parent and teacher ratings of impairment. Intellectual function was measured using the WISC-IV
(53) .
ADHD symptoms in parents were measured using the Parent History Questionnaire
(54), which includes all DSM-IV symptom criteria for ADHD, rated on a 4-point Likert scale (ranging from 0 for “never or rarely” to 3 for “very often”). Separate standardized counts for ADHD symptoms recalled from childhood and ongoing in adulthood, computed using age and gender norms, were used for all analyses. Although the Parent History Questionnaire provides metrics by which to assign categorical diagnoses for both childhood and adult ADHD, we felt that a true diagnosis of ADHD would require a clinical assessment of the adults that was beyond the scope of our study. Community comparison adults completed brief demographic and health history questionnaires.
Measurement of Inhibition
All participants completed the stop-signal task
(21) as a measure of inhibitory control. This task is used to calculate stop-signal reaction time (SSRT), a measure of the latency of the inhibitory process corrected for individual reaction time. Longer SSRT reflects poorer inhibition. The task was presented in four blocks of 24 trials; 18 trials were go trials without stop signals, and the other six included a stop signal. The go stimuli appeared with equal frequency in each block; stop signals were presented randomly and with equal frequency for the right and left hand response. Accuracy, probability of inhibition, and SSRT in each block were examined to validate task compliance. Only participants with valid SSRT scores were included in the sample.
After a complete description of the study, written informed consent and verbal assent were obtained from all participants. The entire research protocol was approved by the Research Ethics Board of the Hospital for Sick Children.
Statistical Analyses
In keeping with previous evidence
(55), age and SSRT score were correlated in the children (r=–0.45, df=175, p<0.01) but not in the adults (r=0.06, df=192, p>0.05). Therefore, covarying for age in analyses that included child and parent data was preferred over the use of age-corrected SSRT score residuals. SSRT score was also correlated with IQ in the children (r=–0.22, df=170, p<0.01). Although the necessity of controlling for IQ in the study of neurocognitive outcomes in children with neurodevelopmental disorders is debatable
(56), IQ was included as a covariate in all analyses that included child data. All analyses were conducted using SPSS, version 15 (Chicago, SPSS).
Generalized linear modeling (GLM) with a normal regression model, identity link function, and robust estimators was used to evaluate the relationship of inhibitory ability between parents and their children. Child and parent data were matched, with parent data repeated in the case of siblings, the robust estimators controlling for the lack of independence within the data set. Univariate analysis of variance was used to compare inhibitory control in parents and adult comparison subjects. GLM with a normal regression model, identity link function, and maximum-likelihood estimators was used to examine the relationship between parental SSRT scores and parental ADHD symptoms. Maximum-likelihood estimators are appropriate in this analysis because the parental data are independent: symptom scores and SSRT scores are not correlated or cross-correlated between mothers and fathers. Group differences in inhibitory control among children with ADHD, their unaffected siblings, and comparison children were evaluated using GLM with a normal regression model, identity link function, and robust estimators. All statistical tests required significant omnibus tests of model fit in advance of examination of factor effects. GLM hypothesis tests provide unstandardized beta weights, standard errors, and significance levels. Since the beta values are unstandardized, they are not comparable across models and are not reported.
Results
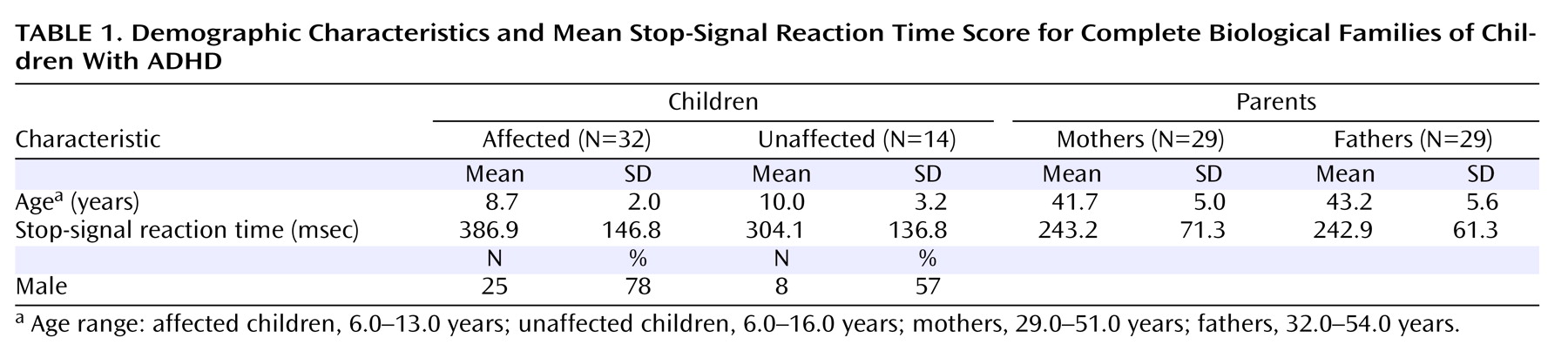
In the children, SSRT score was significantly predicted by age (p<0.001) and mean parental SSRT score (p=0.009) but not by sex, number of symptoms, or number of symptoms in the parent, whether assessed retrospectively in childhood or in adulthood. To determine whether the influence of age or parental SSRT score differed in affected compared with unaffected children, the analysis was repeated with these interactions specified. The interaction of mean parental SSRT score and diagnosis was not a significant predictor of child SSRT score, nor was the interaction of age and diagnosis. When we analyzed only cases in which both parents participated (N=46), no change in the pattern of results was observed (
Table 1 ).
The GLM including child SSRT score as the dependent variable and child age, maternal SSRT score, and paternal SSRT score as predictors indicated that child SSRT score was significantly predicted by age (p<0.001) and by paternal SSRT score (p<0.001) but not by maternal SSRT score. The influence of neither materal SSRT score nor paternal SSRT score on child SSRT score varied by child sex or child diagnosis.
The parents of children with ADHD had poorer inhibitory control relative to the comparison adults (F=24.6, df=1, 190, p<0.001). Mean parental SSRT score was not significantly predicted by age, sex, or number of symptoms, whether measured retrospectively in childhood or in adulthood.
Independent of group differences in IQ, affected children had significantly longer SSRT scores than comparison children (p<0.01). Unaffected siblings had mean SSRT scores intermediate to but not significantly different from those of affected children and comparison children (
Table 2 ). SSRT score was influenced by child age (p<0.001) but not child sex or number of symptoms.
Discussion
This study provides novel lines of evidence supporting the utility of inhibitory control ability as an index of shared genetic risk in ADHD. Parents’ inhibitory control ability significantly predicted the ability of their children, regardless of symptom severity or diagnosis in either generation. We observed covariance across the full measure of inhibitory control in an ADHD sample, not just within the impaired range, which constitutes an important extension of previous research. Furthermore, we found evidence of parent-of-origin effects in this shared risk, with children’s inhibitory control ability influenced more by paternal ability than by maternal ability.
As a marker indexing a limited aspect of genetic risk for the disorder as a whole, an endophenotype should be present in the genetic relatives of affected individuals, independent of symptom severity or diagnostic status. Unaffected siblings showed an inhibition deficit intermediate to those of ADHD children and healthy comparison children, replicating previous findings. Moreover, parents of children with ADHD had a deficit in inhibitory control relative to comparison adults, independent of current symptoms or their history of childhood ADHD. The oft-demonstrated sensitivity and specificity of inhibition to ADHD were also shown in this study by evidence of a greater deficit in ADHD children relative to comparison children. Taken together, our findings replicate and extend evidence supporting motor response inhibition as a cognitive endophenotype in ADHD and suggest avenues for future research.
ADHD is common and highly heritable, but phenotypic and etiological heterogeneity has complicated the search for causal factors. Considerable progress relative to many other heritable disorders notwithstanding, molecular studies of ADHD have been limited by inconsistent findings, low power, and sample selection difficulties. Attempts to reduce heterogeneity in advance of molecular studies have often resulted in the grouping of probands on the basis of behavioral commonalities such as comorbidity
(57 –
59) or subtype
(60,
61) . Properly validated endophenotypes are much more likely to reflect the activity of underlying causal genes. Through their developmental influence on neural substrates, genes influence cognitive processes. Cognitive deficits are clearly evident in ADHD, and most researchers agree that these deficits reflect the disorder’s underlying neural substrate. Thus, cognitive deficits are prime endophenotype candidates in ADHD.
Motor response inhibition meets many of the proposed criteria for a valid endophenotype and has already proved useful in refining the search for genetic risks in ADHD. The presence of an inhibitory deficit in unaffected relatives may further augment the statistical power of genetic linkage and association studies because of its increased prevalence compared with the disease entity, its suitability for quantitative trait analyses, and its ability to clarify affected versus unaffected status in relatives. If inhibitory capacity reflects aspects of the pathophysiology of the disorder
(25,
62,
63), it may also provide a window into neurobiological risk mechanisms not confounded by other etiological factors or clinical sequelae.
Evidence of a parent-of-origin effect in the transmission of inhibitory capacity is also important for future molecular studies. Imprinted, sex-linked, or sex-limited genes are the most common causes of such effects. Our findings suggest that genes on the sex chromosomes are not the source of this effect, since the prediction of child inhibitory ability by maternal and paternal ability did not vary as a function of child sex, although we await replication of this finding in healthy comparison families or with a larger ADHD sample containing more girls.
Many imprinted genes influence brain development and behavior
(64,
65) . Taking imprinting effects into account can substantially improve linkage detection for both discrete and continuous traits
(66) ; ignoring imprinting in molecular analyses severely impairs detection of linkage and may erroneously exclude relevant regions in genome-wide approaches
(66) . Recent evidence also suggests that perturbations of imprinting may be one of the means by which environmental factors influence ADHD risk
(67) .
The limitations of this study include the lack of familial relationship between the child and adult comparison groups and incomplete family triads. Although neither of these factors would be expected to have an influence on the comparison of inhibition in case versus comparison subjects, either factor might influence the interpretation of the regression model assessing the influence of parental measures on child SSRT score. Incomplete family participation is fairly common in familial studies of ADHD, usually because of missing fathers
(15), and this study is no exception. Replication including complete healthy comparison families would help extend the present findings and support quantitative trait analyses using population samples. Future studies should also include full clinical assessments for adults.
Endophenotypes have aided the study of several other conditions in medicine and psychiatry
(68 –
70) . Such findings signal the promise of endophenotypes to better identify and characterize the nature of the genetic contributions to complex disorders. In ADHD, motor control inhibition has overwhelming evidence to support its use in molecular studies of genetic etiology.