Gamma-aminobutyric acid (GABA) is the main inhibitory neurotransmitter of the CNS. While this neurotransmitter plays a major role in the modulation of virtually all cognitive and behavioral processes, clinical studies have particularly highlighted the role of the GABA-ergic system in the central modulation of anxiety and stress responses (
1,
2). To elucidate the mechanisms underlying the antianxiety effects of agents that enhance GABA-ergic neurotrans-mission, a substantial preclinical literature has accumulated, which characterizes the effects of environmental stress on GABA-ergic function in experimental animals. These effects have proven to be complex, since acute stressors can either increase or decrease brain GABA levels, depending on the type of stress, duration of stress, and brain region examined (
3–8). In humans, only indirect evidence exists to support the involvement of GABA in stress response. Reduced GABA plasma concentrations, reduced brain GABA levels as determined by magnetic resonance spectroscopy (MRS), and reduced prefrontal GABA type A (GABA
A)-benzodiazepine receptor complex have consistently been reported in pathological anxiety states and stress-related disorders (
9–12). In both humans and rodents, GABA receptor antagonists generally exert anxiogenic effects, while GABA receptor agonists (e.g., benzodiazepines) and drugs that enhance GABA transmission reduce anxiety and stress responses during exposure to stressors or threats (
13). However, due to the limited availability of noninvasive techniques for assessing GABA-ergic function, the direct effects of acute stress on the central GABA-ergic system have not been studied directly in humans.
The goal of this study was to examine the effects of acute psychological stress on prefrontal GABA concentrations. Subjects underwent two 32-minute GABA scan sessions on different days. One session included an 8-minute well-established threat-of-shock period to induce anxiety, and the other session did not include a threat-of-shock period (control condition). Prefrontal GABA concentrations were determined using a recently developed MRS method with a 3 Tesla General Electric magnetic resonance imaging (MRI) scanner (General Electric, Milwaukee) and a homogeneous resonator coil that allowed for reliable determination of GABA levels with a temporal resolution of 8 minutes. Since stress reduces GABA levels in the medial prefrontal cortex in rat models (
6), we hypothesized that the prefrontal GABA concentration would decrease following acute psychological stress in humans.
Results
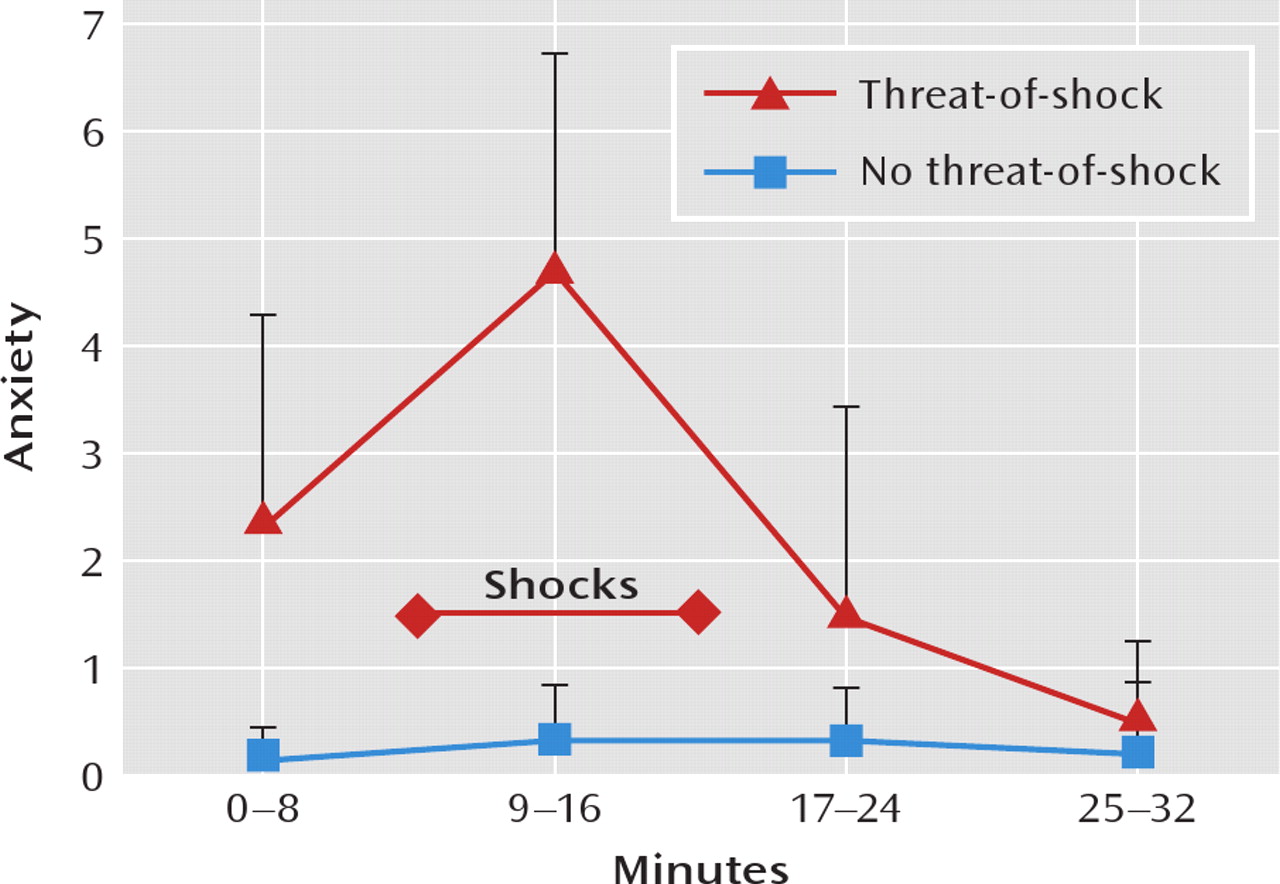
Figure 1 shows the course of self-reported anxiety during the two scan sessions. Subjective anxiety levels were significantly higher during the threat-of-shock session than in the control session (F=29.78, df=1, 67, p<0.0001). In the threat-of-shock session, anxiety levels increased from the first block (safe condition) to the second block (threat condition, p=0.0004) and then decreased from the second to the third block (safe condition, p=0.0003). In the control session, anxiety levels did not differ among blocks.
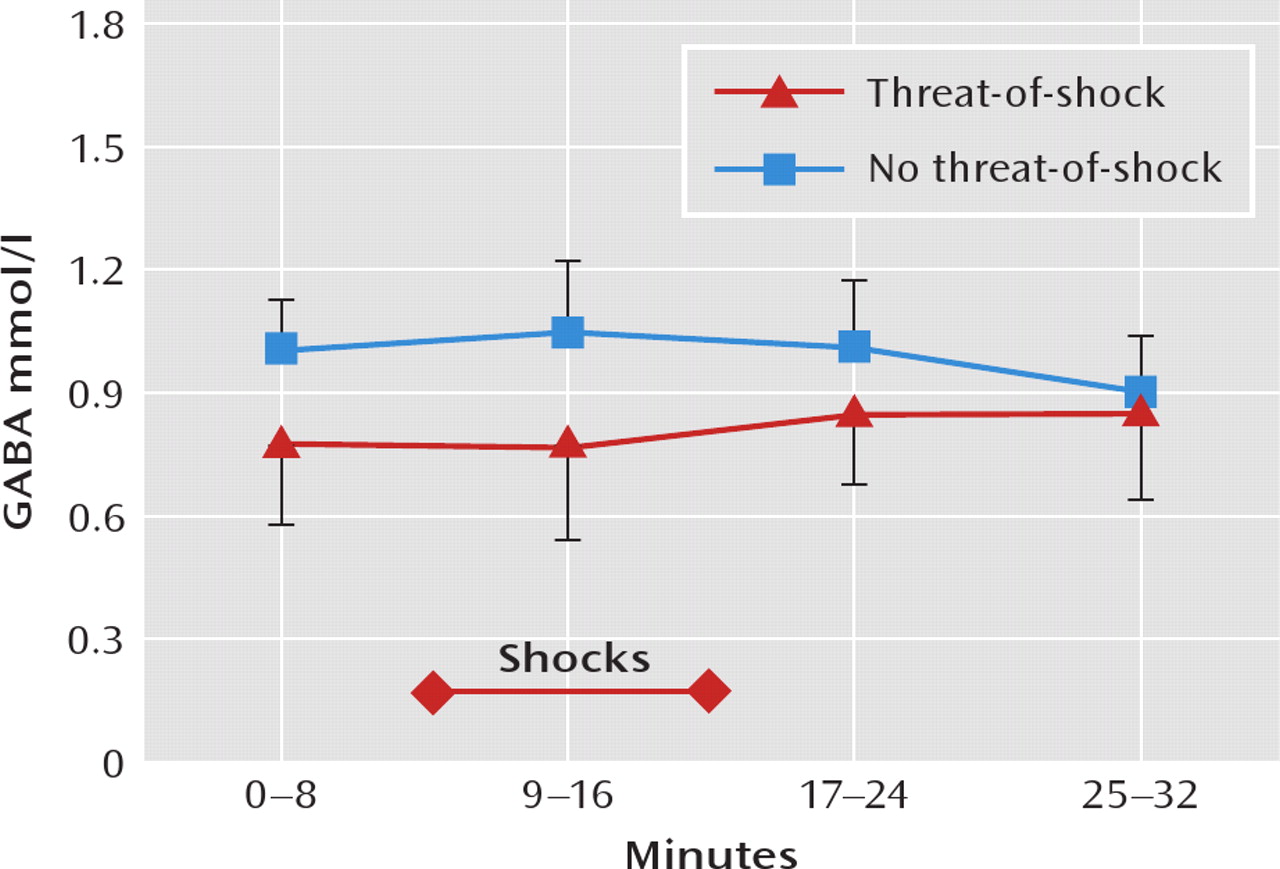
As shown in
Figure 2, GABA levels were significantly lower in the threat-of-shock session than in the control session (F=19.02, df=1, 67, p<0.0001), and the interaction between session type and time was significant (F=9.57, df=1, 67, p=0.003). The difference in GABA concentration between sessions was significant for the first scan block (0–8 minutes, p=0.02), the second block (9–16 minutes, p=0.009), and the third block (17–24 minutes, p=0.004) but was not significant for the fourth block (25–32 minutes). Self-reported anxiety levels and GABA concentrations were inversely correlated (r=–0.31, p=0.005). When including session type (threat-of-shock versus no-threat-of-shock) as a covariate in a model regressing the GABA level on anxiety, the GABA level was no longer associated with anxiety (F=0.81, df=1, 68, p=0.37).
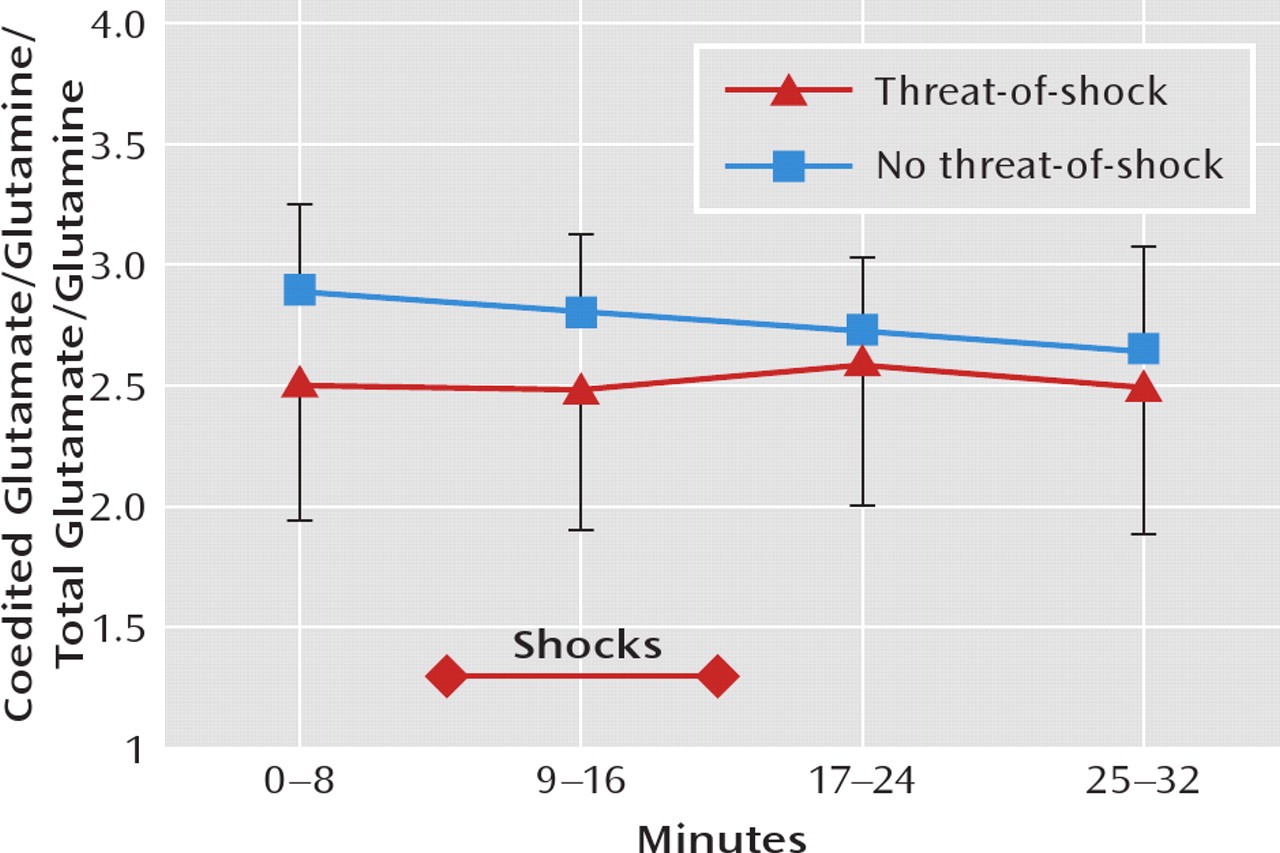
The coedited glutamate/glutamine levels, which are proportional to the total glutamate/glutamine level (
Figure 3 [also see reference
1]), did not differ significantly between the threat-of-shock session and the control session, and there was no session type-by-time interaction. Similarly,
N-acetyl-aspartate and choline levels measured over the entire scan session (32 minutes) did not differ between the threat-of-shock session and the control session (
N-acetyl-aspartate: 9.92 mmol/liter [SD=0.60] versus 9.53 mmol/liter [SD=0.60]; choline: 1.55 mmol/liter [SD=0.16] versus 1.56 mmol/liter [SD=0.17]).
Discussion
This study examined changes in prefrontal GABA concentration in 10 healthy volunteers during a threat-of-shock condition versus a nonthreatening control condition. As expected, the threat-of-shock condition induced robust elevations of subjective anxiety. This was associated with a concomitant reduction in prefrontal GABA concentration.
This change in GABA concentrations between the threat-of-shock session and the control session appeared relatively specific, insofar as they were not associated with significant alterations in the concentrations of N-acetylaspartate, choline-containing compounds, or coedited glutamate/glutamine levels. This observation suggests that the reduction in medial prefrontal cortex GABA concentrations during anticipatory anxiety was not attributable to nonspecific confounds such as head movement, which would cause subtraction errors in the edited spectra and reduce the coupling between the head and radiofrequency resonator coil. Such artifacts would have significantly altered signals of residual water and coedited glutamate/glutamine levels. The measured signals from N-acetyl-aspartate and choline-containing compounds would have also been affected. It is noteworthy that the GABA-editing pulse used in this study has a “top hat” frequency profile around GABA-H3 at 1.89 ppm, with the transition band in the metabolite spectrum from 2.77 ppm to 1.99 ppm. The flat portion of the spectrum reaches from 1.99 ppm to 0.43 ppm, rendering the edited detection of GABA insensitive to the effects of small head movements.
It also appears unlikely that the administration of electric shocks directly contributed to the reduction in GABA levels during the threat-of-shock session. At least 20 minutes elapsed between the sample shocks and the initiation of scanning in block one. In addition, reductions in the prefrontal cortical GABA concentration prior to the threat-of-shock session possibly reflected the effect of anticipatory anxiety. Finally, previous studies reported increased GABA concentrations following electroconvulsive therapy in depressed humans (
22), and no change in GABA was found during electrical stimulation of the forepaws of anesthetized rats (
25).
This study demonstrated a down-modulation of the pre-frontal cortical GABA concentration associated with acute psychological stress and a significant inverse correlation between self-reported anxiety levels and GABA concentrations. A striking feature of the results is that a substantial decrease (nearly 18% averaged over the entire 32-minute scan period) was observed using a relatively large voxel (3×3×2 cm
3). These data are noteworthy, implying that a cortical region that encompasses areas that have been implicated in the brain mapping literature pertaining to anxiety-associated regional blood flow changes observed by positron emission tomography measurements in the pre-frontal cortex (
26,
27) is involved. Brain GABA levels reflect the dynamic balance between GABA synthesis, reuptake, and degradation regulated at the level of glutamate decarboxylase and GABA-transaminase. A decrease in GABA concentration could result from decrease in GABA synthesis or through an increase in GABA degradation by GABA transaminase intracellularly within the mitochondria (
28).
Studies in experimental animals showed consistently that stress modulates GABA release, although the direction of change has varied across brain regions and types of stress. In rats, acute restraint stress increased GABA efflux in the baso-lateral nuclei of the amygdala, whereas efflux remained unchanged in the central nucleus of the amygdala (
29). In the hippocampus, in contrast, forced swimming in 25°C water resulted in a 70% decrease of extracellular GABA concentration, while forced swimming in 35°C water caused a transitory increase in hippocampal GABA (
8). These data suggest that acute stress (i.e., forced swimming in cold water) exerted an effect on GABA concentrations in the rat hippocampus similar in direction to the effect of acute stress we observed in the human medial prefrontal cortex. Exposure to chronic stress has been associated with decreased GABA release (
30,
31). Although we are not aware of preclinical studies that are directly comparable to our study in humans, because of the relatively slow time resolution of our measurements we cannot exclude the possibility that the acute psychological stress we used caused a transitory increase in extracellular GABA followed by more persistent reduction in total GABA concentration in the prefrontal cortex associated with GABA metabolism.
However, the extant data pertaining to the regulation of GABA suggest that the reduced GABA levels in the medial prefrontal cortex under acute stress may reflect a down-regulation of GABA synthesis. At the molecular level, it is well known from neurochemical studies that GABA-transaminase is likely to be saturated with respect to GABA levels under physiological conditions (
32). Therefore, a possible change in GABA degradation is unlikely to account for the observed reduction in GABA concentration. GABA is synthesized in the presynaptic terminals and cell bodies of GABA-ergic interneurons from glutamic acid by the action of glutamate decarboxylase. Although a nearly signifi-cant decrease was observed for the coedited glutamate/ glutamine H
2 peak at 3.8 ppm, a large body of evidence in the literature has shown that there is no definite correlation between alterations in cortical GABA and glutamate/ glutamine concentrations (e.g., reference 33). Taken together, it is suggested that the observed decrease in GABA concentration is likely related to a rapid down-regulation of glutamate decarboxylase associated with acute psychological stress. Glutamate decarboxylase activity and GABA concentrations are modulated during a variety of physiological and pathological conditions, including acute stress (
6,
13). Previous kinetic studies performed on both animals and humans have also shown that GABA undergoes rapid turnover (
34–36). Estimates based on these studies and enzyme kinetic parameters suggest that the rate of change in GABA synthesis is sufficiently rapid to account for the decrease in total GABA concentration observed in this study.
Studies using in vivo microdialysis (e.g., 37) have shown a strong positive correlation between GABA release and total GABA concentration, which appears attributable to a calcium-independent nonvesicular mechanism mediated by rapid reversal of GABA transporters (
38). Thus, the reduction in total prefrontal cortical GABA concentrations as determined by MRS appears to indicate that extracelluar GABA decreases during acute psychological stress. The preclinical literature suggests that this change is most likely associated with a remarkably rapid pre-synaptic modulation of GABA-ergic input in response to acute psychological stress. It might be speculated that the net effect of this reduction in GABA concentrations would be to reduce the overall inhibitory influence of GABA on neural circuits involved in responding to stress or threat. This hypothesis would be compatible with human functional neuroimaging data, which demonstrate that neurophysiological activity normally increases in the medial prefrontal cortex during anxiety provocation or acute stress (
39). The molecular mechanism and functional significance of this reduced inhibitory effect of acute psychological stress in relation to impaired GABA-ergic function in anxiety disorders (
40) merit further investigation.
{kind=link}
{kind=link}
{kind=link}

