Introduction
Major depressive disorder remains a great challenge for clinicians and researchers looking for a window through which to view the inner workings of this common and devastating syndrome. For a disorder affecting nearly 16% of the U.S. population over the course of their lifespan, our knowledge of the fundamental pathophysiology of depression is frustratingly incomplete. The observation that the prevalence is increasing among younger individuals (
1,
2), coupled with the increasingly global scope of the disease (
3), suggests an urgent need for a full-spectrum plan for identifying and ameliorating depression risk factors.
One small window offered by nature is perhaps the most elemental: the human genome. Advancing technology is making it increasingly routine to measure every component of this finite and bounded risk factor for major depression at the level of the individual patient. We already know that the genome contributes to the liability to major depression. Studies of families confirm the notion that major depression is often enriched in families, and studies of twins suggest that this familial aggregation is due to genetic factors (
4). Two studies in this issue of the
Journal (
5,
6) focus their analyses on the crucible of genetics, the family, to extend our knowledge of the genetic contribution to major depression.
In the work by Breen et al. (
5), the investigators assembled a set of 971 sibling pairs in which both members of the pair had two or more episodes of major depressive disorder. Participants were recruited in five European countries and the United States from sites participating in the Depression Network (DeNT) study (
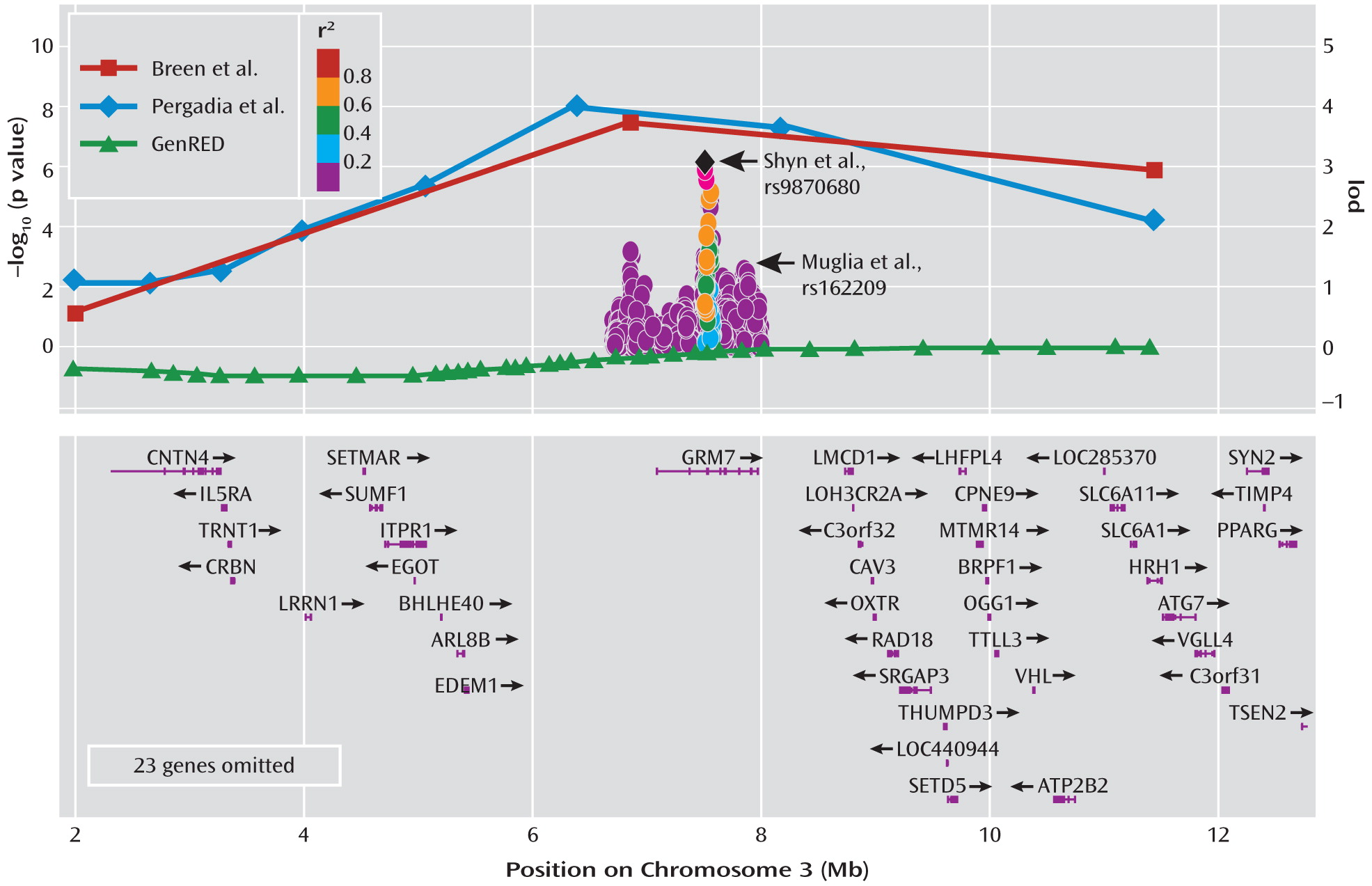
7). Patients were screened to exclude those experiencing bipolar symptoms and psychosis. The cases were graded for degree of severity during the period of peak intensity in each participant's worst two episodes using an impairment item from the diagnostic assessment instrument, resulting in concentric groups of subjects with recurrent major depressive disorder, severe recurrent major depressive disorder, and very severe recurrent major depressive disorder. The authors genotyped the samples for polymorphic markers to determine the parental origin of the chromosomes. They then carried out a statistical operation that determines whether a segment of a chromosome is shared between affected siblings. By chance, siblings should share both copies of any chromosome about 25% of the time and one copy of a chromosome about 50% of the time. Statistically significant deviation from this expectation suggests nonrandom sharing and thus constitutes genetic linkage, which can be presented in the form of a lod (logarithm of odds) score. Breen et al. determined that a lod score of 3.53 represents a statistically significant result based on the number of statistical tests they carried out, and they report that a single region of the genome meets this threshold. The marker they identified resulted in a lod score of 4.0, which they find is equivalent to a p value of 0.015 when adjusted for the genome-wide scale of their experiment. This strongest marker occurs about 500,000 base pairs upstream of a gene called
GRM7, which encodes a protein known as the metabotropic glutamate receptor 7 (mGluR7). Because of the poor resolution of genetic linkage approaches, there are likely to be dozens, if not hundreds, of genes in the region in addition to
GRM7. This finding occurred only in the group with severe recurrent major depression, and not in the sibling pairs concordant for very severe recurrent major depression, a finding ascribed to the smaller numbers in the latter group. The region did not show appreciable linkage in the authors' earlier published analysis of the first 497 sibling pairs (
8).
To validate their findings, Breen et al. then utilized available genotype data from a published genome-wide association study (GWAS) of 1,594 screened control subjects and a set of 2,960 individuals with major depression (
9) overlapping with their linkage sample. Unfortunately, their analysis of ∼1,900 single-nucleotide polymorphisms (SNPs) in the chromosome 3 region did not yield an association that withstood correction for multiple comparisons. While it is possible that this nonreplication is due to the original finding being a false positive, there are other plausible explanations. First, the case-control association sample may not have been large enough to see the expected level of association, although it should have the statistical power to reliably detect a SNP allele conferring an odds ratio of 1.3, which would mean that the odds of major depression occurring in an individual with that SNP allele would be 1.3 times higher than in someone without it. Second, it may be that the linkage approach was better suited to detecting the effect of rare mutations or copy number variants conferring risk in the chromosome 3 region. In other words, if the many sibships that were analyzed harbored many uncommon, even family-specific, risk variants across the same region, the linkage approach would pick up such a cluster of rare variants. This would not necessarily be the situation for the case-control replication sample, where the authors were looking for common risk variants shared by the case subjects. We have a familiar situation, where a seemingly significant result is not replicated and we are left to debate whether the approach is fundamentally incorrect or whether we just do not have sufficient data.
The article by Pergadia et al. (
6) does not start a fire, but does add some smoke. Pergadia et al. utilize a similar approach, using a linkage analysis of 220 sibling pairs affected with a lifetime diagnosis of DSM-IV major depressive disorder, with the families derived from Australian and Finnish twin registries focusing on probands with lifetime cigarette smoking. After genotyping with highly polymorphic markers, the authors report genome-wide significant findings for a marker on chromosome 3 for the Australian component of the sample, which at 187 sibling pairs comprised most of the subjects. The strongest single finding occurred with a lod score of 3.7, again with the nearest gene in the very broad linkage region being
GRM7. Adding more DNA markers and refining the analysis increases the lod score in the region. When the authors examined the smaller Finnish sample, they found no overlap in findings. Together, these articles offer an atypical result for a linkage study of a psychiatric phenotype—linkage not only to the same chromosome but to the same general chromosomal region. While the resolution of linkage cannot tell us the precise location of the signal, the location of both genome-wide significant peaks in the same region is promising.
These articles highlight how geneticists often address challenges they face while seeking to understand psychiatric disorders. A strength of both studies derives from their use of families as the source of their subjects. Using affected sibling pairs may increase the likelihood that individuals with shared genetic risk factors are being studied, as opposed to population-based studies, where the presence of sporadic cases bearing less genetic loading may be found. Similarly, the use of the recurrent major depression phenotype in the study by Breen et al. focuses on a form of major depression shown to be highly familial (
10), enriching the study with individuals for whom genetic factors may be more contributory.
An important requirement for genetic studies is the replicability of findings, which has been a particular problem for linkage-based studies (
11), where it is rare to see replication across samples for complex traits like major depression. The fact that Breen et al. and Pergadia et al. have primary findings that converge on a single chromosomal region suggests that this region may hold great promise, but the findings require additional rigorous proof. The fact that replication samples within each of the studies did not replicate the primary finding is cause for concern. The nonreplication suggests that the main effect may be subtle and obscured by other factors, such as the use of a population-based replication sample in the Breen et al. study or the use of a Finnish sample to replicate the Australian findings in the Pergadia et al. study. Similarly, it is difficult to say that the studies precisely replicate each other, as Pergadia et al. focused on major depression without reference to recurrence or severity, which were the critical aspects of the findings by Breen et al.
The weaker aspects of both articles are related to considerations of phenotype. The study by Breen et al. did not produce significant findings for recurrent major depression and was able to report stronger results only when the authors applied a metric of severity. Even with the severity criterion, Breen et al. found linkage for severe recurrent major depression and not for very severe recurrent major depression, which the authors explain is due to the limited power of the latter analysis. While there is evidence that increasing severity may be related to a more heritable form of major depression (
12), there does not appear to be a consistent approach used by researchers that would allow cross-study comparisons and true replications by other groups.
Perhaps most problematic is the observation that a number of other linkage studies of major depression did not find linkage in the chromosome 3 region, including the large family collections focused on recurrent major depression (
13–
15) or major depression alone (
16). If the small sample used by Pergadia et al. can detect linkage in the chromosome 3 region, it would seem reasonable to think that much larger samples would handily detect the same signal. We should not overinterpret the temporal coincidence of similar findings as being a higher level of evidence. For example, several studies of schizophrenia published over a short period suggested linkage to chromosome 13q (
17–
19), generating much interest and leading to an intense search for a candidate gene (
20) that has proven difficult to confirm (
21). Had the findings of Breen et al. been published in parallel with any of the above major depression linkage studies instead, we would be left to ponder the importance of divergent results.
The Pergadia et al. sample was recruited for a cigarette-smoking phenotype, not for major depression, so their analyses are complicated by the inability to disentangle possible shared susceptibility loci between major depression and smoking. This is not an abstract issue, as other studies using the reverse approach, recruitment for major depression but analysis for smoking, also showed linkage (
22) and association (
23) between smoking-related phenotypes and the same region of chromosome 3. The twin literature supports a complex genetic relationship between major depression and smoking (
24,
25), and we do not know the extent of smoking in the DeNT sample analyzed by Breen et al.
Where do we place these findings in the context of major depression genetics? Until several years ago, many genetic studies of major depression were focused on the examination of hypothesis-driven candidate genes, with very few reproducible findings (
26), or on linkage studies of families with multiple cases of major depression (
11). While family-based studies hold great promise, there is no consensus that increased linkage evidence is a cost-effective way to move the field forward, and it is thus unlikely that funding agencies will invest in new collection and analysis of major depression pedigrees. Nevertheless, much larger family-based samples might ultimately be needed to deal with the seemingly random linkage findings generated by numerous small samples that detect genetic effects that happen to be stochastically enriched in individual samples. In much the same way, GWASs of major depression have revealed a number of new and intriguing associations, although there is little overlap between the individual studies (
27).
Notably, both Breen et al. and Pergadia et al. report that their single-point analysis maximized in a chromosomal region containing the
GRM7 gene, which has gained interest from results of other major depression studies and suggests some convergence of genetic evidence. Recent GWASs of major depression have brought attention to this gene. First, in a report of a GWAS of 1,514 case subjects and 2,052 control subjects of Swiss or German origin, secondary examination of SNPs in major depression candidate genes extracted from the literature revealed a p value of 0.0001 for association between recurrent major depression and a SNP in the last intron of
GRM7 (
28). A subsequent GWAS and meta-analysis of 3,957 major depression case subjects and 3,428 control subjects from the Sequenced Treatment Alternatives to Relieve Depression (STAR*D), Genetics of Recurrent Early onset Depression (GenRED) (
29), and Genetic Association Information Network–MDD (GAIN-MDD) (
30) data sets demonstrated a region of association within
GRM7, with the strongest SNP finding having a p value of 1.11×10
–6and an odds ratio of 1.19 (
31).
Figure 1 shows the striking overlap in findings between the two linkage studies published in this issue and the published GWAS reports.
It is critical to point out that the association results described above do not necessarily explain the linkage findings of Breen et al. and Pergadia et al. The genetic effects of the observed associations from the GWAS would lead to weak penetrance and segregation in families and therefore could not generate the linkage signals observed in the sibling pairs. Even if the linkage signals were coming from the GRM7 gene region, they cannot come from common variants with such small genetic contributions and would have to be due to variants different from those identified by GWAS, as discussed above. Thus, the linkage and GWAS evidence described here represent different types of evidence that are not directly comparable but still do seem to highlight a novel region of the genome.
Subsequent major depression GWAS reports have not reported strong associations with
GRM7 (
9,
32–
34). A meta-analysis of more than 9,000 major depression case subjects and 9,000 control subjects currently under way as part of the Psychiatric GWAS Consortium (
35) will allow the largest available sample to be tested for
GRM7 association. Interestingly,
GRM7 has been noted to be associated in other GWASs, including for the neuroticism-anxiety scale of the Zuckerman-Kuhlman Personality Questionnaire in individuals with bipolar disorder (
36). Rare copy number variants occurring within
GRM7 have been described in mood disorder patients (
37), and a modest association to bipolar disorder (p=0.0001) was noted for a
GRM7 SNP in the Wellcome Trust Case-Control Consortium bipolar disorder GWAS (
38).
mGluR7 is a highly conserved presynaptic autoreceptor expressed widely in the brain that inhibits neurotransmitter release and is thought to be a sort of low-affinity brake on glutamate overstimulation. The finding of a connection between
GRM7 and major depression is exciting, as there is a growing body of research showing a relationship between mGluR7 function and the action of antidepressants and mood stabilizers. For example, in an animal model the mGluR7-specific agonist AMN082 was found to decrease immobility in standard experimental paradigms like the forced swim test and tail suspension tests, whereas these antidepressant-like effects were blocked when
Grm7-deficient mice were used (
39). AMN082 also blocks in vivo acquisition of fear-potentiated startle and facilitates extinction of conditioned fear in animal models (
40), while prolonged citalopram treatment in rats was associated with reductions in immunoreactivity for mGluR7 in frontal cortex and hippocampus (
41). Chronic treatment of rat hippocampal neuronal cultures with valproate or lithium regulates a specific micro-RNA that in turn influences
Grm7 gene expression (
42). Finally, deletion of
Grm7 in mice was found to result in animals showing dysregulation of the hypothalamic-pituitary-adrenal axis (
43) and significantly decreased anxiety- and depression-like behavior in behavioral tests like the elevated plus maze, forced swim test, and tail suspension test (
44).
There are several lessons to be drawn from the two articles. First, GRM7 gains additional support as an exciting gene with great biological plausibility as a focus for additional study and an obvious target for the development of novel therapeutics. Parallel studies in human genetics, basic neuroscience, and clinical psychopharmacology will need to further investigate this possibility. However, some caution should be generated by these findings, especially with regard to claims about finding a “depression gene.” The popular press has often overhyped previous findings that ultimately do not provide consistent associations.
A second lesson lies in how we view replication of findings versus a chance meeting of data sets. To be able to evaluate genetics studies correctly, we have to be able to distinguish between the two situations. In the first, researchers pursue a specific strategy for replicating their findings with the goal of reporting the eventual outcome, whether it supports replication or not. Both articles nicely present their work in this manner, and the replication samples within each study did not replicate the initial finding. In the second situation, there is an unplanned convergence of independent results that may constitute a novel and important finding (as the authors argue) or may represent a type of publication bias in which the studies are reported together since they both happened to be positive. The convergence between the two linkage studies published in this issue was coincidental and not supported by earlier samples of larger size, making their impact less obvious.
A third important lesson is a reminder of how our understanding of the way depression operates in families can provide tangible results. We know that the presence of major depression across multiple generations predicts the development of widespread psychopathology in children (
45), making possible early effective interventions for individuals at high risk (
46). Similarly, treatment of depressed mothers provides substantial benefit to their children (
47), who have an elevated risk for psychopathology. It makes great sense for us to continue deepening our understanding of the role that genetic factors play in depressed families, from further genetic linkage studies like those described here to large-scale genome sequencing studies for identifying the diverse genetic variants that contribute to this common and tragic illness.