Some progress has been made in determining the genetic architecture of late-onset Alzheimer’s disease. The association of late-onset Alzheimer’s disease with the ε4 variant of apolipoprotein E (
APOE) is well established (
1). More recently, replicated genome-wide associations of late-onset Alzheimer’s disease with genetic variation in
CLU,
CR1, and
PICALM have been reported (
2,
3). Ultimately, these detected associations must be related to specific neurobiological processes leading to Alzheimer’s disease in order for them to translate into meaningful clinical predictors or therapies. Our current understanding of the neurobiology of Alzheimer’s disease suggests that disease causation is dependent on a sustained period of pathological accumulation of β-amyloid protein (see reference
4 for a review). The initial accumulation of β-amyloid occurs prior to clinically detectable cognitive impairment, setting off downstream events, including generation of hyperphosphorylated tau, gray matter volume reductions, and synapse loss (
5,
6). It is measures of these downstream effectors that appear to change most rapidly during phases of disease characterized by more rapid cognitive decline (
4).
Conceptually, genetic variation may thus be linked to cognitive change in Alzheimer’s disease in any of several ways. Genetic variations that increase the production of pathological β-amyloid protein, including mutations and duplications of the β-amyloid precursor protein gene (
APP), and mutations in presenilin 1 and 2 (
PSEN1 and
PSEN2), all lead to an earlier age at disease presentation (
7).
Similarly, APOE ε4 alleles, which reduce β-amyloid clearance (8), are associated with earlier age at disease presentation (
9). Alternatively, one might postulate that genetic variation whose primary role is to interact with the generated or accumulated β-amyloid so as to modify its downstream effects would influence the rate of overt cognitive decline. To date, no such genetic variations have been definitively identified. However, psychotic symptoms in Alzheimer’s patients define a genetically determined behavioral phenotype (
10) that is strongly associated with a more rapid rate of cognitive decline (
11–
13).
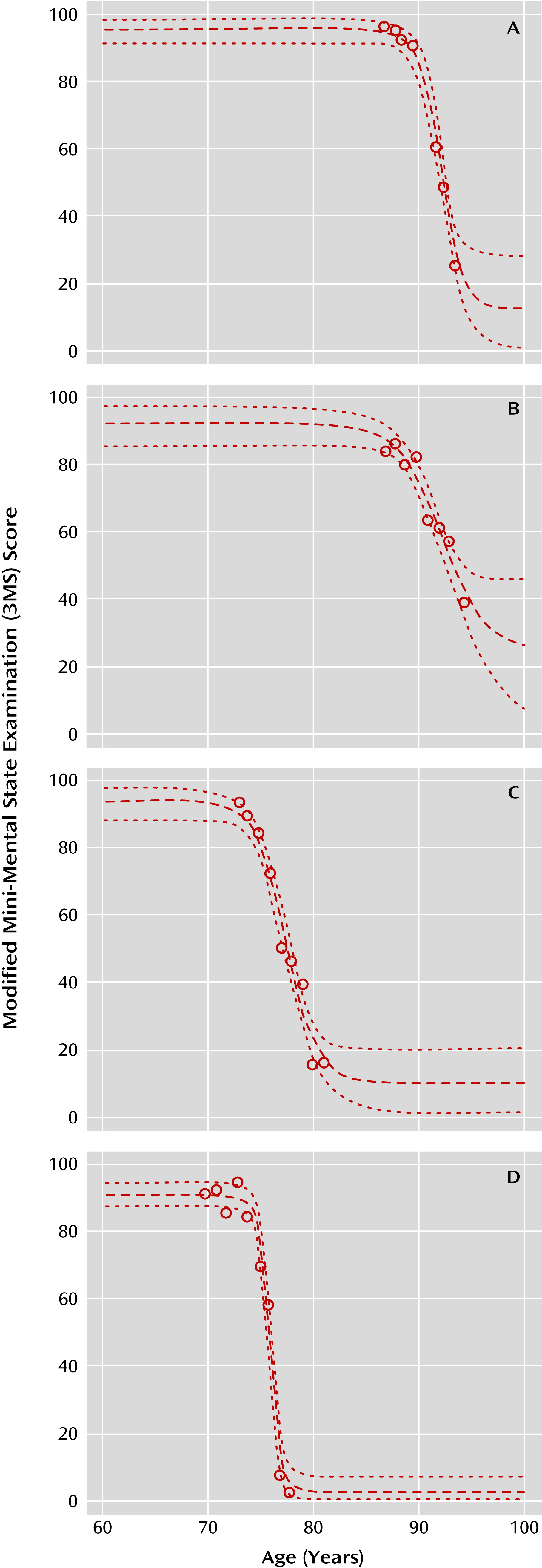
It would therefore be useful for interpreting the nature of a genetic variation’s relationship to Alzheimer’s disease risk to be able to measure cognitive trajectories in a way that would allow testing of genetic associations with both age and rate of cognitive decline. Intuitively, it can be seen that an individual traversing the complete course of Alzheimer’s disease will have a period prior to disease onset of relatively stable cognitive test performance, followed by a period of declining performance, until reaching a period of scores asymptotically approaching a minimum score. Such a trajectory is readily approximated by a four-parameter logistic curve, but several obstacles exist in practice. These include the fact that few longitudinal data sets gather complete information on all stages of the cognitive trajectory for individual Alzheimer’s patients, although the picture may be complete for the group as a whole. Additionally, it is not uncommon for elderly individuals to have poor cognitive test performance in a given session as a result of factors unrelated to Alzheimer’s disease, such as concomitant illness, poor sleep, or medication effects. Trajectory modeling must not be overly sensitive to these outlier values.
Discussion
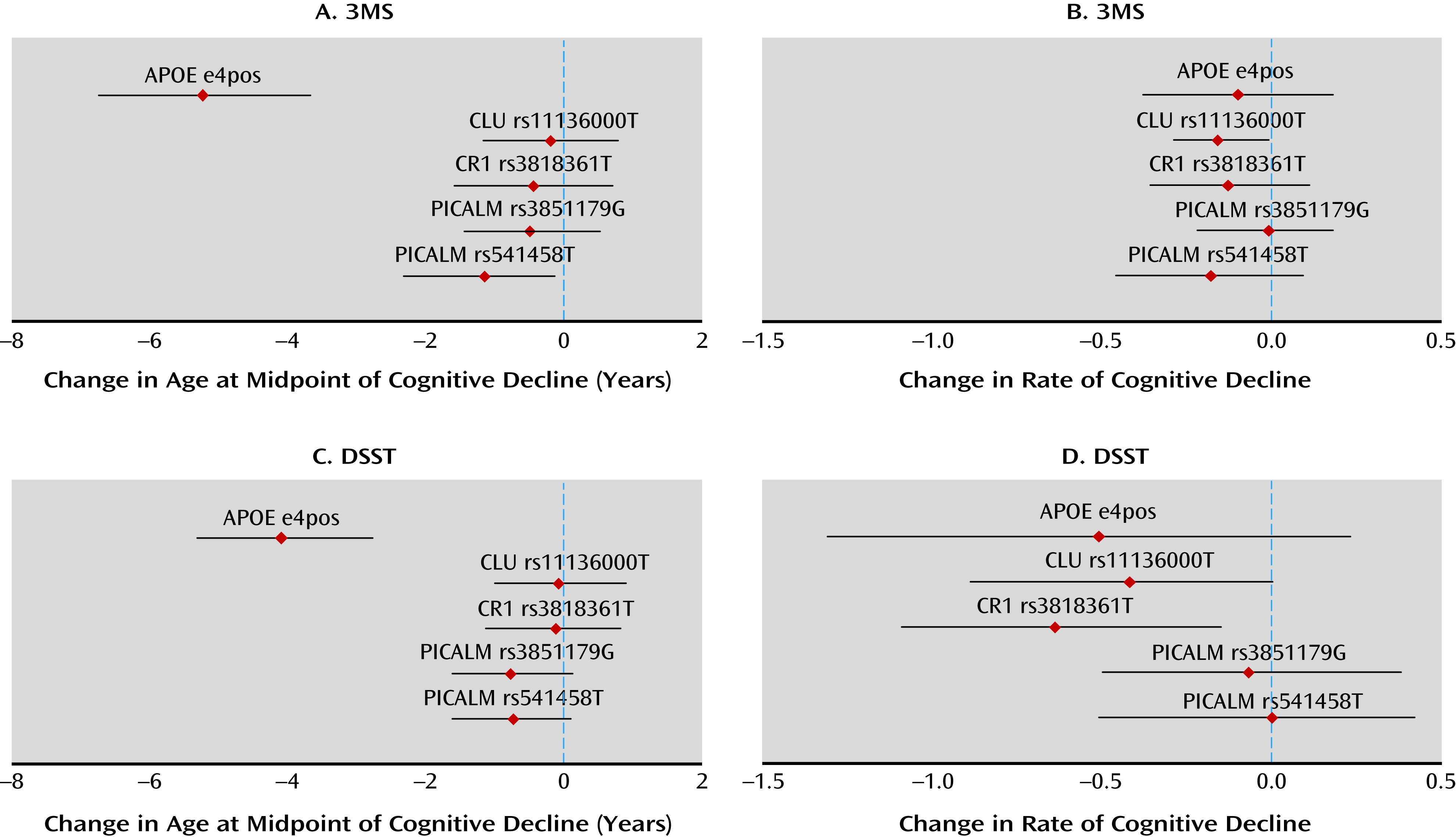
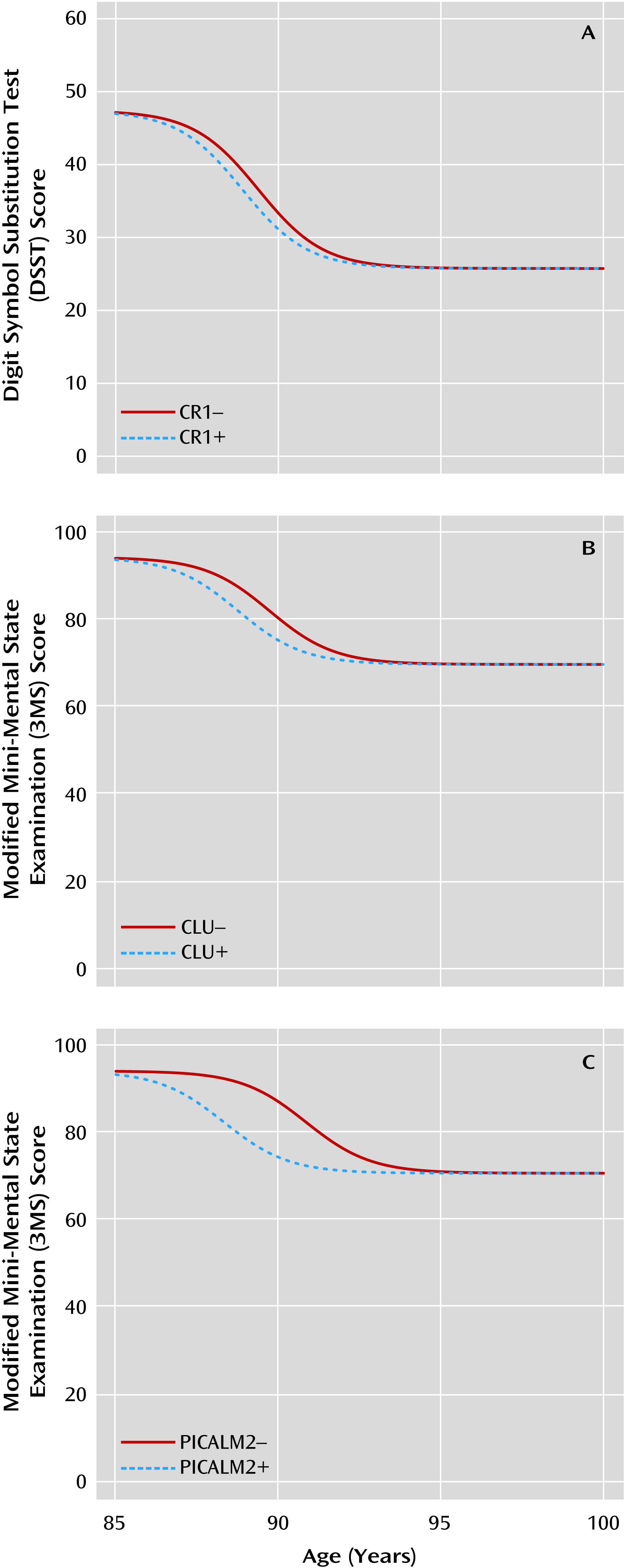
We found that a nonlinear model implemented with a Bayesian hierarchical analytic approach generated robust fits to the observed cognitive trajectories of elderly subjects. Validating our approach, APOE ε4 alleles were associated with a lower age at midpoint of cognitive decline, and psychosis was associated with a higher rate of cognitive decline. We then implemented our model to test whether SNPs associated with an increased risk for late-onset Alzheimer’s disease alter cognitive trajectory after accounting for the effects of APOE and demographic factors. We found that rs11136000 in CLU and rs3818361 in CR1 were associated with more rapid cognitive decline. rs541458 in PICALM was associated with an earlier age at midpoint of cognitive decline, although this latter association did not persist in an additive model designed to correct, in part, for multiple testing. For all SNPs except CLU SNP rs11136000, the allele conferring an adverse cognitive trajectory was the same as that associated with Alzheimer’s disease in previous GWAS.
We used a nonlinear model that allowed for censoring at the maximum and minimum values. This choice was based on a common property of cognitive tests, which often have defined maximum and minimum scores. As a result, prior to disease onset individuals may sustain maximal scores, while in end-stage disease sustained minimal scores may be present. Such observations cannot be fitted adequately with a trajectory derived from a linear or quadratic model, although multiple linear fits (e.g., as applied in change-point models), may be used to approximate subsets of points within a longitudinal cognitive trajectory (
23). Other psychometric properties of cognitive tests also contribute to nonlinearity. Many tests are admixtures of easier, moderately difficult, and difficult questions, not necessarily in equal proportion. As a consequence, scores may change little during a given phase of cognitive decline, for example, during early illness if there are few difficult questions. Alternatively, scores may show greater change during another phase, such as moderate disease, if there are proportionally more questions of moderate difficulty. Many global cognitive tests, including the 3MS, follow just such a pattern (
24). In contrast, the DSST requires repeating the same cognitive function and is scored based on how many times it is successfully completed within a timed interval. However, the overall difficulty of the task results in nonlinearity in moderate to severely impaired individuals, supporting the use of a nonlinear model to fit these data (
25). Determining detection of SNP effects would be enhanced by the use of cognitive measurements that demonstrate more consistent linear measurement properties across levels of disease severity.
We found that rs11136000 in
CLU was associated with more rapid cognitive decline, although the risk for more rapid decline was conferred by the allele opposite to that previously associated with Alzheimer’s disease (
2,
3,
20,
26). Although rate of cognitive decline and presence of Alzheimer’s disease are not identical, it seems unlikely that a causal allele would confer different directions of risk for these two outcomes. Several authors have argued that significant allelic association in opposing directions is statistically unlikely (
27,
28) and probably reflects true association via one of several underlying mechanisms (
27–
30).
CLU encodes the protein clusterin, which is expressed at elevated levels in the brains of individuals with Alzheimer’s disease and can serve to prevent fibrillization of β-amyloid and inhibit complement activation (
31). In individuals with mild cognitive impairment and Alzheimer’s disease, elevated plasma clusterin levels have been found to be correlated with more rapid cognitive decline, although plasma clusterin concentrations were not associated with genetic variants in
CLU, including rs11136000 (
32).
We found that rs3818361 in
CR1 was associated with more rapid cognitive decline.
CR1 is a complement receptor expressed in cerebral cortex. There is emerging evidence that the complement cascade contributes to targeting synapses for elimination during development and in neurodegeneration via astrocyte-mediated opsonization with the complement component 3 fragment, C3b (
33).
CR1 supports clearance of opsonized targets by its high affinity for C3b (
34). Recent evidence indicates that the reported associations of genetic variation in
CR1 with Alzheimer’s disease may arise from linkage disequilibrium between these variants and a low copy repeat that codes for a
CR1 isoform with an additional C3b binding site (
35). It is not currently known whether
CR1 contributes to synapse elimination in Alzheimer’s disease.
Finally, we found that rs541458 in
PICALM was associated with an earlier age at midpoint of cognitive decline, although it should be noted that this association did not persist in our additive model.
PICALM encodes the phosphatidylinositol-binding clathrin assembly (Picalm) protein, an essential factor in clathrin-mediated endocytosis (
36). Recent evidence indicates that Picalm is primarily expressed in vascular endothelial cells in human brain (
37), where it may affect the clearance of β-amyloid from brain. For example, rs541458 in
PICALM is significantly associated with CSF levels of β-amyloid 42 (
38). This interpretation would be congruent with a mechanism by which variants that result in overproduction or overaccumulation of β-amyloid most strongly affect age at cognitive decline.
These associations of
CLU, CR1, and
PICALM were detected in models that accounted for demographic variables and the effect of
APOE ε4. This finding stands in contrast to a recent analysis of several cohorts, including the Cardiovascular Health Study cohort, which found that rs11136000 in
CLU and rs3851179 in
PICALM did not add to the prediction of Alzheimer’s disease onset beyond demographic and
APOE effects (
20). This may indicate that there is increased power available in using a cognitive trajectory, rather than a dichotomous diagnosis, to identify SNP associations with longitudinal outcome in subjects at risk for Alzheimer’s disease. Of interest, we found that genetic variation in
PICALM was associated with lower age at midpoint of decline in models that included
APOE. This is somewhat surprising given recent evidence that
APOE and
PICALM genotypes are correlated, resulting in substantial reduction in the association of
PICALM SNPs with Alzheimer’s disease risk after accounting for
APOE status (
26). Unlike
PICALM, associations of
CLU and
CR1 with Alzheimer’s disease risk did not display confounding with
APOE genotype (
26); thus, it is not surprising that they demonstrated detectable effects after controlling for
APOE ε4 alleles.
We used a Bayesian approach to fit cognitive trajectories. The principal advantage of Bayesian methods over classical (e.g., mixed-model) analyses is that calculation of posterior distributions is typically straightforward even for complex models. However, Bayesian methods are often computer intensive, creating a potential limitation of our approach; it may not be practical for sequential screening of large numbers of SNPs. Another potential limitation in any model fitting is inadequate model convergence. In Bayesian hierarchical models, completely uninformative prior distributions may result in invalid (improper) posterior distributions. The standard solution to this problem is to incorporate a small amount of prior information to create “weakly informative” prior distributions. In clinical settings such as the ones we modeled, this is simple and effective, because we can put reasonable limits on parameters—for example, we know that for our subjects the age at midpoint of cognitive decline is between 30 and 130 years. The utility of these weak prior distributions can be seen in the fidelity of our trajectory fits to the observed data. Finally, an important modification to our Bayesian implementation was the use of a t-distribution rather than a normal distribution to model deviations of measurements from trajectory means. This approach substantially reduced the effects of occasional outlier values, a not uncommon occurrence in our observed data (see Figure S1 in the online
data supplement).
Other possible limitations to our findings should be considered. We selected for testing a limited number of SNPs based on replicated evidence of genome-wide significant association in GWAS. Other SNPs in these genes, which did not themselves reach genome-wide significant association with Alzheimer’s disease risk, may nevertheless be associated with trajectory of cognitive decline. The same may be true for genetic variation in other genes, including a number of SNPs that have been identified as demonstrating genome-wide significant association in GWAS of Alzheimer’s disease. Determining a trajectory requires a minimum number of observed measurements. We chose as inclusion criteria the presence of four measurements on the 3MS and DSST tests. This requirement may have excluded some individuals with more rapid decline, who thus did not complete four tests. Similarly, the individuals who participated in the Cardiovascular Health Cognition Study and were thus available for this analysis were a nonrandom subset of all Cardiovascular Health Study participants, healthier and younger than the parent cohort. Thus, our findings may not fully generalize to the elderly population.
In summary, we developed a Bayesian approach to address several goals for the analysis of cognitive trajectories in aging subjects, including the ability to fit individual as well as group cognitive trajectories; make meaningful descriptive statements about the pattern of change and the variability in that pattern across subjects; develop credible regions for the resultant curves, which include appropriate prediction limits; and evaluate the specific effects of covariates, including genetic covariates, by estimating their impact on trajectory parameters. We validated this approach with two established predictors of different cognitive trajectory parameters, the presence of APOE ε4 alleles and the development of psychosis. We then used our approach to detect effects of recently identified Alzheimer's risk alleles on cognitive trajectories. Replication of this approach using other data sets is warranted.