Dopamine Synthesis Capacity in Patients With Treatment-Resistant Schizophrenia
Publication: American Journal of Psychiatry
Abstract
Objective
Elevated presynaptic striatal dopaminergic function is a robust feature of schizophrenia. However, the relationship between this dopamine abnormality and the response to dopamine-blocking antipsychotic treatments is unclear. The authors tested the hypothesis that in patients with schizophrenia the response to antipsychotic treatment would be related to the severity of presynaptic dopamine dysfunction, as indexed using [18F]-DOPA uptake positron emission tomography (PET).
Method
Twelve patients with treatment-resistant schizophrenia, twelve patients with schizophrenia who had responded to antipsychotics, and twelve healthy volunteers matched for gender, age, ethnicity, weight, and cigarette smoking underwent [18F]-DOPA PET scanning. [18F]-DOPA influx rate constants (Kicer values) were measured in the striatum and its functional subdivisions.
Results
Patients who had responded to antipsychotic treatment showed significantly higher Kicer striatal values than did patients with treatment-resistant illness (effect size=1.11) and healthy volunteers (effect size=1.12). The elevated [18F]-DOPA uptake was most marked in the associative (effect size=1.31) and the limbic (effect size=1.04) striatal subdivisions. There were no significant differences between patients with treatment-resistant illness and healthy volunteers in the whole striatum or any of its subdivisions.
Conclusions
In some patients with schizophrenia, antipsychotic treatment may be ineffective because they do not exhibit the elevation in dopamine synthesis capacity that is classically associated with the disorder; this may reflect a different underlying pathophysiology or a differential effect of antipsychotic treatment.
Between 20% and 35% of patients with schizophrenia show only a limited response to antipsychotic treatment (1). This treatment resistance is a major clinical problem, impairing the lives of these patients.
The dopamine hypothesis of schizophrenia proposes that dysregulated dopaminergic function underlies the positive psychotic symptoms of the disorder (2). Supporting this hypothesis, eight radiolabeled DOPA positron emission tomography (PET) studies have reported that dopamine synthesis capacity is elevated in schizophrenia, with effect sizes ranging from 0.63 to 1.89 (3). Furthermore, elevated dopamine synthesis capacity predates the onset of psychosis (4, 5) and progressively increases as patients move from the prodromal phase to the first episode of illness (6). Other molecular imaging studies in schizophrenia indicate that both dopamine release and baseline dopamine levels are elevated (7, 8).
There is substantial evidence that the efficacy of antipsychotic drugs is related to dopamine D2 receptor blockade (9). All currently approved antipsychotic medications block dopamine receptors (10), and their relative clinical potency closely parallels their binding and blocking affinity for the dopamine D2 receptor subtype (11). PET studies have demonstrated a significant association between striatal D2 occupancy and short-term clinical response to antipsychotic treatment (12) and have indicated that at least 50% occupancy of D2 receptors is necessary to achieve clinical response (13). Wolkin et al. (14) have investigated D2 receptor occupancy in relation to treatment resistance in schizophrenia. They found almost identical levels of striatal dopamine receptor occupancy in patients who responded to antipsychotic treatment and those with treatment-resistant illness, suggesting that while adequate D2 blockade may be necessary for a therapeutic response, it does not guarantee it.
Dopamine levels have also been linked to treatment response. It has been shown that the higher the level of striatal synaptic dopamine, the better the subsequent response to antipsychotic treatment (7). However, to our knowledge no study has examined dopamine synthesis capacity specifically in relation to treatment resistance.
The limited data available suggest that patients who do not respond to current dopamine-blocking treatments may have a lower level of [18F]-DOPA uptake than those who respond to dopaminergic blockers (7, 15). However, it could also be argued that patients who do not respond to antipsychotic treatment have an exceptionally hyperactive dopaminergic system that is not blocked by current antipsychotics. We sought to address this issue by using [18F]-DOPA PET to compare dopamine synthesis capacity in patients who have shown a good response to antipsychotic treatment with those who have shown a poor response, as well as with healthy comparison subjects.
Method
The study protocol was approved by the research ethics committee of the Institute of Psychiatry, King’s College London, and permission to administer radioactive substances was granted by the Administration of Radioactive Substances Advisory Committee, United Kingdom. All participants gave written informed consent to participate after receiving a full description of the study.
We recruited two groups of patients, defined according to their response to antipsychotic treatment. All met DSM-IV criteria for schizophrenia, paranoid subtype, determined by using OPCRIT (the Operational Criteria Checklist) (16). The group with treatment-resistant illness (treatment-resistant group; N=14) comprised patients who met modified Kane criteria for treatment resistance (17). All of these patients had previously received at least two sequential antipsychotic trials, each of at least 4 weeks’ duration at a daily dose of 400–600 mg of chlorpromazine equivalents, but continued to have persistent psychotic symptoms, which was defined as having a rating of at least moderate severity on one or more items on the positive symptom subscale of the Positive and Negative Syndrome Scale (PANSS) (18), having a total PANSS score ≥75 (19), and having a score <59 on the Global Assessment of Functioning (corresponding to at least moderate functional impairment) (20). The group of patients who responded to antipsychotic treatment (responder group; N=12) comprised patients who met the Remission in Schizophrenia Working Group criteria for treatment remission (21). These patients scored ≤3 on all items of the PANSS (corresponding to mild severity or no symptoms) and had not experienced a symptomatic relapse in the 6 months prior to the study. All patients were recruited from the South London and Maudsley NHS Trust. A group of healthy comparison subjects (N=12) with no previous or current history of psychiatric illness (as assessed by the Structured Clinical Interview for DSM-IV Axis I Disorders and the Structured Clinical Interview for DSM-IV Personality Disorders) and no family history of psychosis were recruited through advertisements in the press. The groups were matched for age, gender, ethnicity, weight, and smoking. Patients in the treatment-resistant and responder groups were matched, in addition, for duration of illness and for antipsychotic dosage (in chlorpromazine equivalents).
All patients were receiving antipsychotic medication other than clozapine at the time of scanning. Two patients in the treatment-resistant group had previously been treated with clozapine, but it had been discontinued because of side effects without reaching therapeutic blood levels. Adherence to medication was determined by measuring antipsychotic drug serum levels and by reviewing pharmacy and medical records. Patients were excluded if there was evidence of nonadherence at any point in the 6 months prior to the scan or if serum levels were not at adequate levels. Exclusion criteria for all groups were pregnancy (all women received a pregnancy test prior to scanning), contraindication to imaging, history of neurological or active medical illness or head injury, or substance abuse or dependence. All patients received a urine drug screen prior to scanning and were excluded if it was positive for illicit substances.
One patient was excluded from the treatment-resistant group because he did not complete the scanning, and one patient in the treatment-resistant group withdrew from the study. The data analysis was therefore restricted to 12 patients in the treatment-resistant group, 12 patients in the responder group, and 12 healthy volunteers.
PET Protocol
Participants were instructed to fast and to refrain from caffeine, tobacco, and alcohol for at least 12 hours before scanning. One hour before the start of each scan, all subjects received 150 mg p.o. of carbidopa, a peripheral aromatic acid decarboxylase inhibitor, and 400 mg p.o. of entacapone, a peripheral catechol-O-methyltransferase inhibitor, to increase specific signal detection, as these compounds reduce the formation of radioactively labeled metabolites that may cross the blood-brain barrier and thus confound the measurements.
PET imaging data were acquired on a Siemens/CTI ECAT HR+ 962 PET scanner (Erlangen, Germany) in three-dimensional mode, with an axial field of view of 15.5 cm. Participants were positioned in the scanner with the orbitomeatal line parallel to the transaxial plane of the tomograph. Head position was monitored via laser crosshairs and video camera. The initial transmission scan was followed by administration of approximately 180 MBq of the radiotracer [18F]-DOPA, a radioactive analog of l-dopa, as a bolus intravenous injection over 30 seconds. Emission data were obtained as 26 frames of increasing duration over 90 minutes (comprising a 30-second background frame, four 60-second frames, three 120-second frames, three 180-second frames, and finally fifteen 300-second frames). In addition, structural MRI was conducted to exclude intracranial abnormalities. No gross abnormalities were detected in any participant in a review by a neuroradiologist blind to the subject group.
Image Analysis
Both automated region-of-interest and voxel-based statistical image analyses with the cerebellum as reference region were performed to examine striatal [18F]-DOPA uptake, as previously described by our group (4–6). The region-of-interest analysis (performed by A.D.) included the whole striatum and its associative, limbic, and sensorimotor subregions, delineated as described by Martinez et al. (22). These functional striatal subdivisions reflect the differential striatal-cortical connectivity and the functional organization of the striatum. The limbic striatum, anatomically equivalent to the ventral striatum, receives input from limbic structures such as the hippocampus and amygdala and includes the nucleus accumbens. The associative striatum comprises the precommissural dorsal caudate, precommissural dorsal putamen, and postcommissural caudate and receives input from associative cortical regions, including the dorsolateral prefrontal cortex. The sensorimotor striatum includes the postcommissural putamen and receives projections predominantly from motor and premotor areas (22). The region-of-interest map thus comprised the whole striatum, its subdivisions, and the cerebellum, defined using a probabilistic atlas.
To correct for head movement, the nonattenuated dynamic image was denoised and realigned frame-to-frame to a single reference frame acquired 7 minutes after [18F]-DOPA injection. Next, the transformation parameters were applied to the corresponding attenuation-corrected frames, and the realigned frames were summated to create a movement-corrected dynamic image ready for analysis. The region-of-interest map was then normalized together with an [18F]-DOPA template to each individual PET summation image using SPM5 (www.fil.ion.ucl.ac.uk/spm), which allows region of interest to be placed automatically and without observer bias on individual [18F]-DOPA PET dynamic images. A Patlak graphical analysis was used to calculate striatal [18F]-DOPA influx rate constants (Kicer values) (23) to index striatal dopamine synthesis capacity relative to uptake in the reference region (the cerebellum) for left and right sides combined.
Voxel-based statistical image analysis was performed to independently confirm the results derived from region-of-interest analysis and determine whether there were subregional differences. Parametric maps of the influx rate constants for [18F]-DOPA were constructed from movement-corrected images by using a wavelet-based kinetic modeling approach that increases the signal-to-noise ratio without significantly affecting resolution (see Howes et al. [4]). After normalization of the parametric images, statistical analyses were performed using SPM5, restricted to the striatum using a mask, to examine differences between groups. The results presented were analyzed with correction for multiple comparisons (p<0.05, family-wise error rate) and, in a further sensitivity analysis, without correction.
Statistical Analysis
We conducted preliminary tests to explore homogeneity of variance, regression slopes, normality, and reliable measurements of covariates. The Kolmogorov-Smirnov test confirmed that the data were normally distributed. To determine whether there was an effect of group on striatal Kicer values and on demographic, striatal volume, and clinical data, analysis of variance and independent t tests were performed as appropriate. When there were significant group effects, planned independent t tests were performed using Bonferroni correction for multiple comparisons to examine differences in Kicer values between groups. To assess whether the effect was influenced by medication, an additional analysis of covariance was performed with daily medication dose (in chlorpromazine equivalents) added as a covariate. The independent variable was group, and the dependent variables were the Ki values for the striatum and its three subdivisions. A two-tailed significance threshold of 0.05 was used throughout.
Results
Demographic and clinical characteristics are presented in Table 1, and antipsychotic use is summarized in Table 2. No between-group differences were observed for age, gender, ethnicity, weight, radiation dose received, cigarette smoking, duration of illness, or medication dosage. In addition, there were no differences across the groups for the whole striatal volume or any of its subdivisions.
| Variable | Patients With Treatment-Resistant Illness (N=12) | Treatment Responders (N=12) | Healthy Volunteers (N=12) | |||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | |
| Age (years) | 45.7 | 9.8 | 44.0 | 11.9 | 44.2 | 8.9 |
| Weight (kg) | 80.9 | 19.2 | 83.0 | 20.7 | 80.9 | 23.3 |
| Radioactivity injected (MBq) | 180.0 | 5.5 | 183.6 | 4.3 | 183.4 | 5.6 |
| Specific activity (GBq/μmol) | 0.025 | 0.017 | 0.033 | 0.015 | 0.035 | 0.019 |
| Duration of illness (years) | 16.1 | 8.6 | 16.2 | 10.1 | ||
| Medication dosage (mg/day chlorpromazine equivalents) | 396.1 | 157.5 | 283.9 | 159.14 | ||
| Positive and Negative Syndrome Scale | ||||||
| Total score | 104.3 | 10.6 | 50.7 | 5.8 | ||
| Positive symptom score | 26.2 | 3.8 | 11.9 | 2.4 | ||
| Global Assessment of Functioning score | 47.5 | 3.9 | 67.5 | 4.5 | ||
| Whole striatal volume (mm3) | 15,590.9 | 1,675.2 | 16,424.1 | 1,818.9 | 16,773.6 | 1,870.1 |
| Associative striatal volume (mm3) | 9,644.0 | 1,165.5 | 10,185.0 | 1,164.1 | 10,005.0 | 1,768.2 |
| Limbic striatal volume (mm3) | 1,953.1 | 262.3 | 1,965.5 | 348.0 | 2,013.1 | 269.3 |
| Sensorimotor striatal volume (mm3) | 4,260.2 | 928.6 | 4,189.6 | 513.5 | 4,394.4 | 459.6 |
| N | % | N | % | N | % | |
| Male | 5 | 41.6 | 6 | 50.0 | 5 | 41.6 |
| Smoker | 3 | 25.0 | 4 | 33.3 | 3 | 25.0 |
| Ethnicity | ||||||
| White | 5 | 41.6 | 4 | 33.3 | 6 | 50.0 |
| Black | 6 | 50.0 | 7 | 58.3 | 4 | 33.3 |
| Asian | 1 | 8.3 | 1 | 8.3 | 2 | 16.7 |
a
No significant differences between groups on any variable.
| Patients With Treatment-Resistant Illness (N=12) | Treatment Responders (N=12) | |||
|---|---|---|---|---|
| Antipsychotic | N | % | N | % |
| Olanzapine | 3 | 25.0 | 2 | 16.7 |
| Quetiapine | 2 | 16.7 | 1 | 8.3 |
| Amisulpride | 1 | 8.3 | 1 | 8.3 |
| Risperidone | 1 | 8.3 | 0 | 0.0 |
| Risperidone depot | 2 | 16.7 | 3 | 25.0 |
| Aripiprazole | 0 | 0.0 | 1 | 8.3 |
| Chlorpromazine | 1 | 8.3 | 0 | 0.0 |
| Zuclopenthixol decanoate | 1 | 8.3 | 3 | 25.0 |
| Flupenthixol decanoate | 1 | 8.3 | 1 | 8.3 |
a
No significant differences between groups.
Figure 1 shows the mean dopamine synthesis capacity for the three groups. The analysis of variance identified a statistically significant effect of group on Kicer values for the whole striatum (F=5.4, df=2, 33, p=0.01) and for each of its associative (F=6.7, df=2, 33, p=0.004), limbic (F=4.0, df=2, 33, p=0.03), and sensorimotor subdivisions (F=3.4, df=2, 33, p=0.05). Mean Kicer values are listed in Table 3.
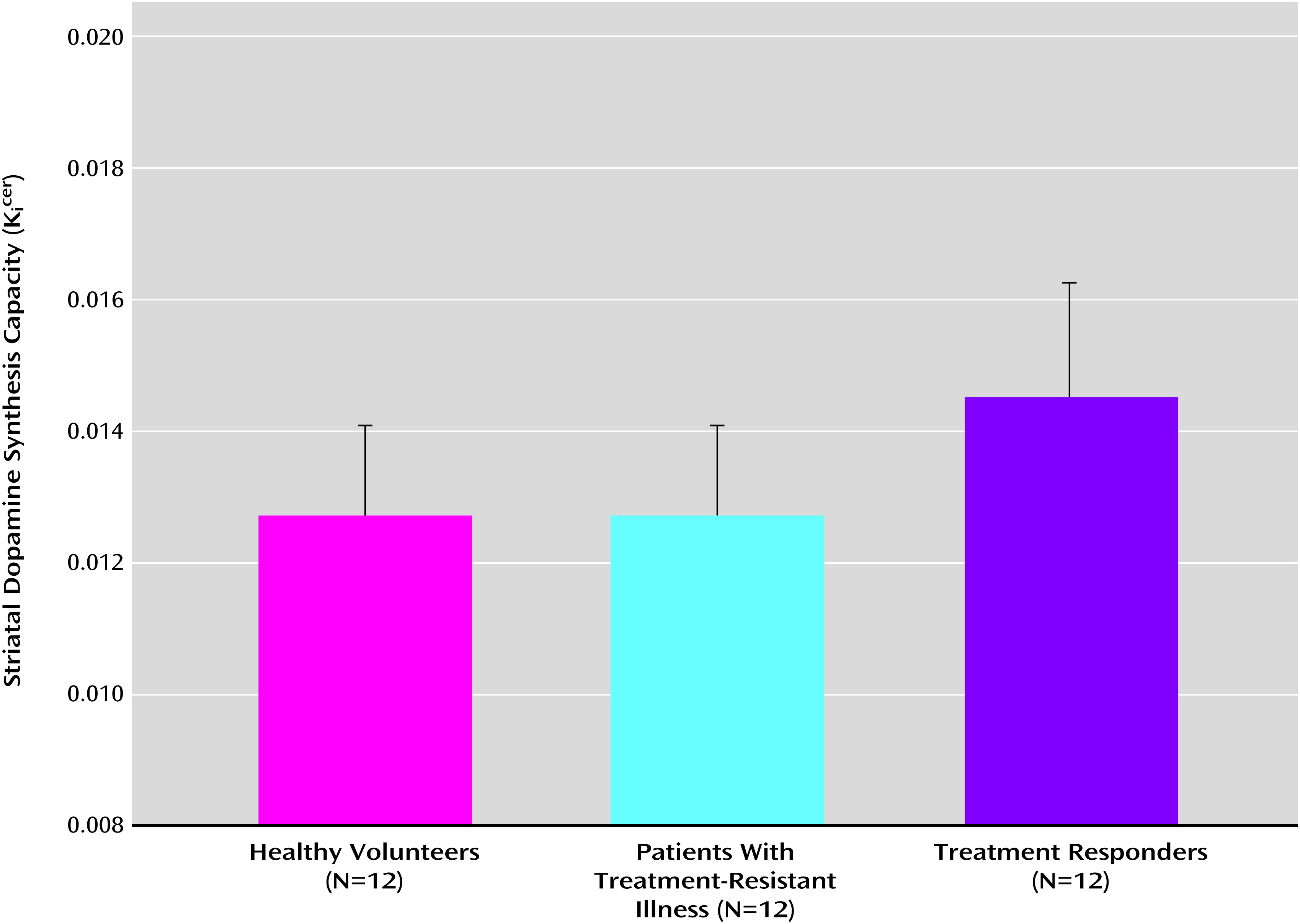
FIGURE 1. Mean Dopamine Synthesis Capacity for the Whole Striatum in Patients With Treatment-Resistant Schizophrenia, Treatment Responders, and Healthy Volunteersa
a The treatment-resistant group showed significantly lower dopamine synthesis capacity than the treatment responders (p=0.02, corrected for multiple comparisons). There were no significant differences between treatment-resistant patients and healthy volunteers. Error bars indicate standard deviation.
| Patients With Treatment-Resistant Illness (N=12) | Treatment Responders (N=12) | Healthy Volunteers (N=12) | Analysis | |||||
|---|---|---|---|---|---|---|---|---|
| Brain Structure | Mean | SD | Mean | SD | Mean | SD | F (df=2,33) | p |
| Whole striatum | 1.3×10−2 | 0.14×10−2 | 1.4×10−2 | 0.18×10−2 | 1.3×10−2 | 0.14×10−2 | 5.38 | 0.01 |
| Associative striatum | 1.2×10−2 | 0.14×10−2 | 1.4×10−2 | 0.15×10−2 | 1.2×10−2 | 0.14×10−2 | 6.71 | 0.004 |
| Limbic striatum | 1.3×10−2 | 0.17×10−2 | 1.5×10−2 | 0.23×10−2 | 1.3×10−2 | 0.17×10−2 | 4.02 | 0.03 |
| Sensorimotor striatum | 1.4×10−2 | 0.2×10−2 | 1.6×10−2 | 0.24×10−2 | 1.4×10−2 | 0.15×10−2 | 3.38 | 0.05 |
Because one of the patients in the responder group had particularly high Kicer values, we repeated the analysis with that subject excluded. The group effect remained significant for the whole striatum (F=4.2, df=2, 32, p=0.02) and its associative subdivision (F=6.4, df=2, 32, p=0.005), but not the other striatal subdivisions.
To assess the effect of antipsychotic medication, the analysis was repeated with the addition of medication dose (in chlorpromazine equivalents) at the time of scanning as a covariate. The effects of group on Kicer values from the whole striatum (F=7.2, df=1, 22, p=0.01) and the associative (F=10.2, df=1, 22, p=0.005) and limbic (F=6.4, df=1, 22, p=0.02) subdivisions remained significant, but there was no longer a significant difference in the sensorimotor subdivision.
Between-Group Comparisons
Treatment-resistant versus responder group.
After adjustment for multiple comparisons, Kicer values were significantly greater in the responder group than in the treatment-resistant group in the whole striatum (p=0.02, corrected; effect size=1.11) and the associative (p=0.008, corrected; effect size=1.31) and limbic subdivisions (p=0.03, corrected; effect size=1.04). There was no significant difference in the sensorimotor subdivision.
Greater Kicer values in the responder group than in the treatment-resistant group were also observed in the corresponding voxel-based analysis, with a peak in the head of the caudate (p=0.039), which lies within the associative subdivision of the striatum (Figure 2). The difference was significant at p<0.05, corrected for multiple comparisons using the family-wise error rate. The treatment-resistant group > responder group contrast showed no significant differences, even at an uncorrected statistical threshold (p<0.05, uncorrected).
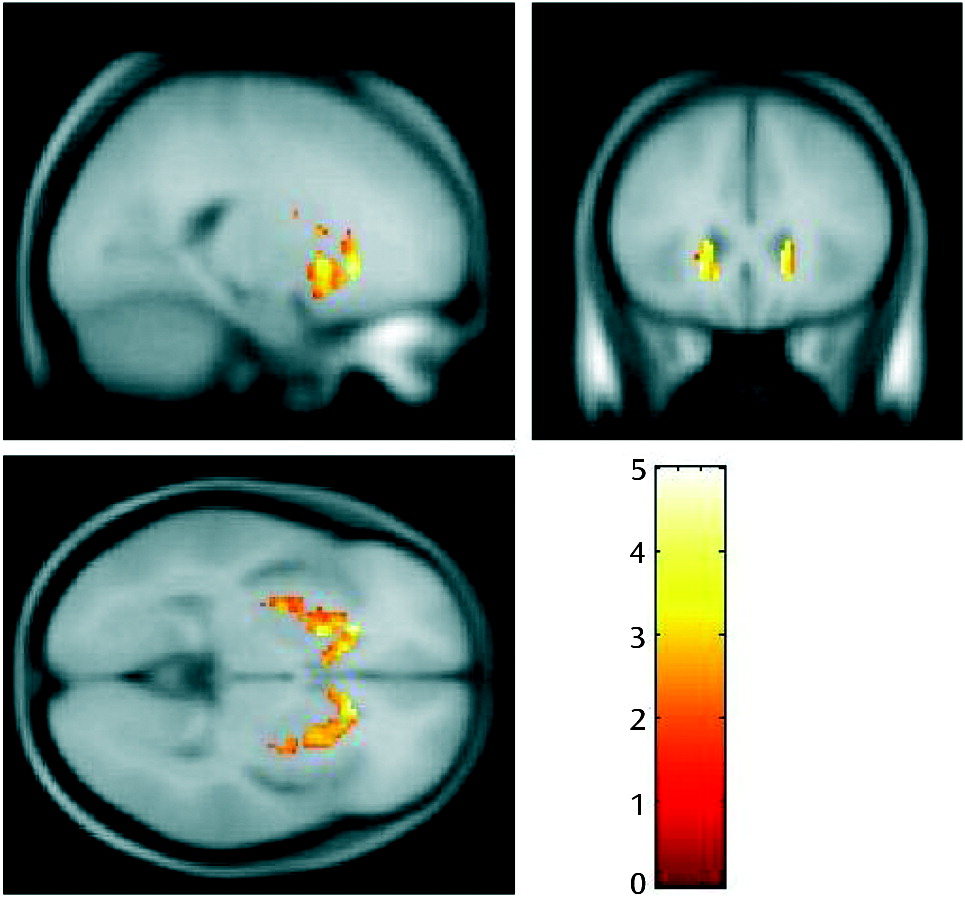
FIGURE 2. Greater Striatal Dopamine Synthesis Capacity in Schizophrenia Treatment Responders Relative to Patients With Treatment-Resistant Illnessa
a The images show increased dopamine synthesis capacity in responders (N=12) relative to patients with treatment-resistant illness (N=12). The most significant increase was in the head of the caudate nucleus (p=0.039, corrected at the family-wise error rate).
Responder group versus healthy volunteers.
Kicer values were significantly elevated in the responder group compared with the healthy volunteer group, after multiple comparison adjustments, in the whole striatum (p=0.02, corrected; effect size=1.12) and the associative subdivision (p=0.01, corrected; effect size=1.24), but not in the limbic or sensorimotor subdivisions. Similarly, the voxel-based analysis revealed significantly greater Kicer values in the responder group compared with the healthy volunteer group, with a peak in the caudate (p=0.037, corrected at the family-wise error rate). The voxel-based contrast of the healthy volunteers with the responder group showed no significant differences, even at an uncorrected threshold (p<0.05 uncorrected).
Treatment-resistant group versus healthy volunteers.
There was no significant difference in mean striatal Kicer values between the treatment-resistant group and the healthy volunteers, in the whole striatum or its subdivisions. This was confirmed with the subsequent voxel-based analysis for the contrast of treatment-resistant group > healthy volunteer group and for the contrast of the healthy volunteer group > treatment-resistant group, even at an uncorrected threshold (p<0.05, uncorrected).
Discussion
To our knowledge, this is the first study to provide direct evidence that dopamine synthesis capacity in schizophrenia is lower in patients with treatment-resistant illness than in those who show a good response to antipsychotic medication. This suggests that treatments that involve the blockade of dopamine receptors may be effective in patients who have an elevation of dopamine synthesis capacity but less useful in patients in whom dopamine synthesis capacity is relatively normal.
Limitations
One potential limitation of our study is that the patients were chronically medicated, which could have influenced presynaptic dopamine synthesis capacity (24). The two patient groups were matched for both the current dosage of medication and the total duration of treatment. The mean daily dose was higher in the treatment-resistant group (reflecting their poor response to treatment), but the difference in dose was not statistically significant, and group differences in Kicer values remained significant after covarying for dose. Another potential limitation is that our patients received various types of antipsychotic drugs that could have differentially affected dopamine synthesis. However, the groups were relatively well matched in terms of generation and type of antipsychotics (see Table 2). Two of our patients, one in the treatment-resistant group and the other in the responder group, were taking amisulpride, which at dosages lower than 200 mg daily may increase dopamine transmission via a preferential blockade of presynaptic D2-like autoreceptors (25). However, as these two patients received much higher dosages—800 mg and 600 mg daily, respectively—than the level for a preferential presynaptic action and were in different groups, it is unlikely that our results were affected. Only one of our patients (in the responder group) was taking aripiprazole, which has the unique property of being a partial dopamine receptor agonist (25). Theoretically, partial agonists should reduce dopamine synthesis capacity, reflected in a lower the Kicer value, although in practice little net effect has been observed during chronic aripiprazole administration in rats (26). As the aripiprazole-treated patient was in the responder group, a reduction in Kicer value would reduce rather than account for the group differences we observed between the responder and treatment-resistant groups. Furthermore, as the Kicer values for this individual were similar to the mean Kicer value for the group, it is unlikely that including the aripiprazole-treated individual had a major effect on the overall results.
As the group sizes were relatively modest, the possibility that the absence of differences in dopamine synthesis capacity between the treatment-resistant group and the healthy volunteers could reflect limited statistical power should be also considered. However, data from previous studies in schizophrenia suggest that the effect size for the elevation in dopamine synthesis capacity measured using the same PET protocol is relatively large (>1) (4, 27), and a formal power calculation indicated that a sample size of 12 per group had 80% power to detect an effect size of >0.7, using a two-group two-sided t test and a significance threshold of 0.05.
Presynaptic Dopamine Synthesis Capacity in Treatment-Resistant Schizophrenia
The lack of an elevation in presynaptic striatal dopamine synthesis capacity in patients with treatment-resistant illness could provide an explanation for the ineffectiveness of antipsychotic treatment in this group. Our findings are in agreement with studies of plasma homovanillic acid in patients prior to treatment with antipsychotics: levels were higher in the responder group than in the treatment-resistant group (15). Furthermore, Abi-Dargham et al. (7) found that higher synaptic dopamine levels, as indexed by D2 receptor occupancy, were associated with a better response to antipsychotic treatment. There is evidence from animal studies that chronic treatment with dopamine-blocking antipsychotics induces D2 receptor up-regulation, which can reduce the efficacy of antipsychotic treatment and may lead to breakthrough dopamine supersensitivity (28, 29). Whether these elevated D2 receptors may then affect dopamine synthesis in patients with treatment-resistant illness is not entirely clear. Our study was cross-sectional and therefore cannot determine whether presynaptic dopamine synthesis capacity was normal in patients in the treatment-resistant group at the onset of their illness, or whether it was initially abnormal but then some change occurred so that the persistent psychotic symptoms were no longer related to a striatal dopamine excess. Kolakowska et al. (30) observed that in most of their patients with treatment-resistant illness, the response to antipsychotic treatment had been insufficient throughout the illness, which led them to conclude that the treatment response was linked to the type rather than the stage of schizophrenia. Establishing whether differences in the severity of dopamine dysfunction predate exposure to antipsychotics will require long-term prospective studies of therapeutic response in patients who are initially medication naive.
Higher Presynaptic Dopamine Synthesis Capacity in Responders
We observed significantly higher presynaptic dopamine synthesis capacity in patients who showed a good response to antipsychotics, with the strongest effect observed in the associative subdivision of the striatum, consistent with previous evidence (4, 5, 31). While the elevation in the responder group is consistent with some previous PET studies in chronic patients (27, 32), there are conflicting reports from other studies in chronic stable patients with schizophrenia that found no significant dopamine elevation (33, 34). These studies, however, did not distinguish specifically between patients with a good response to antipsychotics and those with treatment-resistant illness, which may explain the discrepancy in results. Thus, the studies that found a dopamine elevation could have included predominantly responders, and those that reported no elevation may have included more patients with treatment-resistant illness.
One tentative explanation for the paradoxically high dopamine synthesis capacity in the face of relative symptomatic remission could be that in the context of chronic exposure to D2 receptor blockade, these patients do not have enhanced transmission, because of the ambient postsynaptic D2 blockade. However, it is not clear whether antipsychotic drugs do normalize dopamine synthesis capacity. In one study (35) acute treatment was found to increase dopamine synthesis capacity, although in another study (36) no overall effect was observed. On the other hand, Gründer et al. (37) have reported that longer-term antipsychotic treatment reduces presynaptic dopamine synthesis capacity, and preclinical studies (38, 39) have shown that antipsychotics induce delayed depolarization block of presynaptic dopamine neurons, an effect that is more rapidly induced in a rat schizophrenia model showing increased dopamine neuron activity than in wild-type rats. However, the Gründer et al. study (37) did not include healthy volunteers; thus, while antipsychotics reduced dopamine synthesis capacity, it remains unknown whether antipsychotic treatment normalized dopamine synthesis or not. Our study and some other studies (4, 27), although not all (34), involving antipsychotic-treated patients suggest that dopamine synthesis capacity is not completely normalized by antipsychotic treatment. Finally, the effect size seen in our responder group approximates that observed in previous studies of dopamine synthesis capacity in schizophrenia, including those involving medication-free or medication-naive patients (4). Thus, overall, these findings suggest that medication does not explain the elevation seen in the responder group.
Conclusions
These data indicate that schizophrenia patients whose illness is resistant to antipsychotic treatment have relatively normal levels of dopamine synthesis capacity, compared with levels in patients whose symptoms respond to treatment. This suggests either that patients with treatment-resistant illness start with a different underlying pathophysiology or that antipsychotics have an effect on their dopamine synthesis capacity, albeit one that does not reduce symptoms. Since our study was cross-sectional and comprised a small sample of medicated patients, our results require replication, ideally in prospective studies of large samples of antipsychotic-naive patients. Future studies need to determine the involvement of other neurotransmitters. In particular, evidence that glutamate dysfunction may contribute to the pathophysiology of schizophrenia and to the efficacy of clozapine in patients who have not responded to treatment with stronger D2 antagonists suggests that changes in the glutamate system and its interaction with other neurotransmitters may be an important factor in this subgroup (40).
Acknowledgments
The authors thank the participants and the staff at the Hammersmith Hospital, particularly Hope McDewit, Stephanie McKnight, and Andrew Blyth.
Footnote
Supported by grant U.1200.04.007.00001.01 from the Medical Research Council, U.K.; the National Institute for Health Research Biomedical Research Centre for Mental Health; and a Medical Research Council Strategic Award to Dr. Kapur.
References
1.
Lindenmayer JP: Treatment refractory schizophrenia. Psychiatr Q 2000; 71:373–384
2.
Howes OD, Kapur S: The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophr Bull 2009; 35:549–562
3.
Howes OD, Montgomery AJ, Asselin MC, Murray RM, Grasby PM, McGuire PK: Molecular imaging studies of the striatal dopaminergic system in psychosis and predictions for the prodromal phase of psychosis. Br J Psychiatry Suppl 2007; 51:s13–s18
4.
Howes OD, Montgomery AJ, Asselin MC, Murray RM, Valli I, Tabraham P, Bramon-Bosch E, Valmaggia L, Johns L, Broome M, McGuire PK, Grasby PM: Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry 2009; 66:13–20
5.
Howes OD, Bose SK, Turkheimer F, Valli I, Egerton A, Valmaggia LR, Murray RM, McGuire P: Dopamine synthesis capacity before onset of psychosis: a prospective [18F]-DOPA PET imaging study. Am J Psychiatry 2011; 168:1311–1317
6.
Howes O, Bose S, Turkheimer F, Valli I, Egerton A, Stahl D, Valmaggia L, Allen P, Murray R, McGuire P: Progressive increase in striatal dopamine synthesis capacity as patients develop psychosis: a PET study. Mol Psychiatry 2011; 16:885–886
7.
Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann JJ, Van Heertum RL, Gorman JM, Laruelle M: Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci USA 2000; 97:8104–8109
8.
Laruelle M, Abi-Dargham A, van Dyck CH, Gil R, D’Souza CD, Erdos J, McCance E, Rosenblatt W, Fingado C, Zoghbi SS, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB: Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci USA 1996; 93:9235–9240
9.
Kapur S, Zipursky R, Jones C, Remington G, Houle S: Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry 2000; 157:514–520
10.
Talbot PS, Laruelle M: The role of in vivo molecular imaging with PET and SPECT in the elucidation of psychiatric drug action and new drug development. Eur Neuropsychopharmacol 2002; 12:503–511
11.
Creese I, Burt DR, Snyder SH: Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 1976; 192:481–483
12.
Kapur S, Zipursky R, Jones C, Shammi CS, Remington G, Seeman P: A positron emission tomography study of quetiapine in schizophrenia: a preliminary finding of an antipsychotic effect with only transiently high dopamine D2 receptor occupancy. Arch Gen Psychiatry 2000; 57:553–559
13.
Abi-Dargham A, Laruelle M: Mechanisms of action of second generation antipsychotic drugs in schizophrenia: insights from brain imaging studies. Eur Psychiatry 2005; 20:15–27
14.
Wolkin A, Barouche F, Wolf AP, Rotrosen J, Fowler JS, Shiue CY, Cooper TB, Brodie JD: Dopamine blockade and clinical response: evidence for two biological subgroups of schizophrenia. Am J Psychiatry 1989; 146:905–908
15.
Duncan E, Wolkin A, Angrist B, Sanfilipo M, Wieland S, Cooper TB, Rotrosen J: Plasma homovanillic acid in neuroleptic responsive and nonresponsive schizophrenics. Biol Psychiatry 1993; 34:523–528
16.
McGuffin P, Farmer A, Harvey I: A polydiagnostic application of operational criteria in studies of psychotic illness: development and reliability of the OPCRIT system. Arch Gen Psychiatry 1991; 48:764–770
17.
Conley RR, Kelly DL: Management of treatment resistance in schizophrenia. Biol Psychiatry 2001; 50:898–911
18.
Kay SR, Fiszbein A, Opler LA: The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull 1987; 13:261–276
19.
Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR: What does the PANSS mean? Schizophr Res 2005; 79:231–238
20.
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 4th ed Washington, DC, American Psychiatric Association, 1994
21.
Andreasen NC, Carpenter WT, Kane JM, Lasser RA, Marder SR, Weinberger DR: Remission in schizophrenia: proposed criteria and rationale for consensus. Am J Psychiatry 2005; 162:441–449
22.
Martinez D, Slifstein M, Broft A, Mawlawi O, Hwang DR, Huang Y, Cooper T, Kegeles L, Zarahn E, Abi-Dargham A, Haber SN, Laruelle M: Imaging human mesolimbic dopamine transmission with positron emission tomography, part II: amphetamine-induced dopamine release in the functional subdivisions of the striatum. J Cereb Blood Flow Metab 2003; 23:285–300
23.
Patlak CS, Blasberg RG, Fenstermacher JD: Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab 1983; 3:1–7
24.
Carlsson A, Lindqvist M: Effect of chlorpromazine or haloperidol on formation of 3-methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol (Copenh) 1963; 20:140–144
25.
Miyamoto S, Duncan GE, Marx CE, Lieberman JA: Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry 2005; 10:79–104
26.
Der-Ghazarian T, Charntikov S, Varela FA, Crawford CA, McDougall SA: Effects of repeated and acute aripiprazole or haloperidol treatment on dopamine synthesis in the dorsal striatum of young rats: comparison to adult rats. J Neural Transm 2010; 117:573–583
27.
McGowan S, Lawrence AD, Sales T, Quested D, Grasby P: Presynaptic dopaminergic dysfunction in schizophrenia: a positron emission tomographic [18F]fluorodopa study. Arch Gen Psychiatry 2004; 61:134–142
28.
Samaha AN, Seeman P, Stewart J, Rajabi H, Kapur S: “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci 2007; 27:2979–2986
29.
Ginovart N, Wilson AA, Hussey D, Houle S, Kapur S: D2-receptor upregulation is dependent upon temporal course of D2-occupancy: a longitudinal [11C]-raclopride PET study in cats. Neuropsychopharmacology 2009; 34:662–671
30.
Kolakowska T, Williams AO, Ardern M, Reveley MA, Jambor K, Gelder MG, Mandelbrote BM: Schizophrenia with good and poor outcome, I: early clinical features, response to neuroleptics, and signs of organic dysfunction. Br J Psychiatry 1985; 146:229–239
31.
Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, Hwang DR, Huang Y, Haber SN, Laruelle M: Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry 2010; 67:231–239
32.
Reith J, Benkelfat C, Sherwin A, Yasuhara Y, Kuwabara H, Andermann F, Bachneff S, Cumming P, Diksic M, Dyve SE, Etienne P, Evans AC, Lal S, Shevell M, Savard G, Wong DF, Chouinard G, Gjedde A: Elevated dopa decarboxylase activity in living brain of patients with psychosis. Proc Natl Acad Sci USA 1994; 91:11651–11654
33.
Shotbolt P, Stokes PR, Owens SF, Toulopoulou T, Picchioni MM, Bose SK, Murray RM, Howes OD: Striatal dopamine synthesis capacity in twins discordant for schizophrenia. Psychol Med 2011; 41:2331–2338
34.
Elkashef AM, Doudet D, Bryant T, Cohen RM, Li SH, Wyatt RJ: 6-(18)F-DOPA PET study in patients with schizophrenia: positron emission tomography. Psychiatry Res 2000; 100:1–11
35.
Vernaleken I, Kumakura Y, Cumming P, Buchholz HG, Siessmeier T, Stoeter P, Müller MJ, Bartenstein P, Gründer G: Modulation of [18F]fluorodopa (FDOPA) kinetics in the brain of healthy volunteers after acute haloperidol challenge. Neuroimage 2006; 30:1332–1339
36.
Ito H, Takano H, Takahashi H, Arakawa R, Miyoshi M, Kodaka F, Okumura M, Otsuka T, Suhara T: Effects of the antipsychotic risperidone on dopamine synthesis in human brain measured by positron emission tomography with L-[beta-11C]DOPA: a stabilizing effect for dopaminergic neurotransmission? J Neurosci 2009; 29:13730–13734
37.
Gründer G, Vernaleken I, Müller MJ, Davids E, Heydari N, Buchholz HG, Bartenstein P, Munk OL, Stoeter P, Wong DF, Gjedde A, Cumming P: Subchronic haloperidol downregulates dopamine synthesis capacity in the brain of schizophrenic patients in vivo. Neuropsychopharmacology 2003; 28:787–794
38.
Grace AA, Bunney BS, Moore H, Todd CL: Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci 1997; 20:31–37
39.
Valenti O, Cifelli P, Gill KM, Grace AA: Antipsychotic drugs rapidly induce dopamine neuron depolarization block in a developmental rat model of schizophrenia. J Neurosci 2011; 31:12330–12338
40.
Carlsson A, Waters N, Holm-Waters S, Tedroff J, Nilsson M, Carlsson ML: Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol 2001; 41:237–260
Information & Authors
Information
Published In


History
Received: 31 January 2012
Revision received: 15 May 2012
Accepted: 18 June 2012
Published online: 1 November 2012
Published in print: November 2012
Authors
Funding Information
Dr. Murray has received speakers honoraria from AstraZeneca, Bristol-Myers Squibb, Janssen, Eli Lilly, and Roche. Dr. Kapur has received grant support from or served as an adviser, speaker, or consultant to AstraZeneca, Bioline, Bristol-Myers Squibb, GlaxoSmithKline, Janssen, Eli Lilly, Lundbeck, NeuroSearch, Otsuka, Pfizer, Roche, Servier, and Solvay Wyeth. Dr. Howes has received investigator-led charitable funding or spoken at meetings organized by AstraZeneca, Bristol-Myers Squibb, Janssen-Cilag, and Eli Lilly. Drs. Demjaha and McGuire report no financial relationships with commercial interests.
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBFigures
FIGURE 1. Mean Dopamine Synthesis Capacity for the Whole Striatum in Patients With Treatment-Resistant Schizophrenia, Treatment Responders, and Healthy Volunteersa
a The treatment-resistant group showed significantly lower dopamine synthesis capacity than the treatment responders (p=0.02, corrected for multiple comparisons). There were no significant differences between treatment-resistant patients and healthy volunteers. Error bars indicate standard deviation.
FIGURE 2. Greater Striatal Dopamine Synthesis Capacity in Schizophrenia Treatment Responders Relative to Patients With Treatment-Resistant Illnessa
a The images show increased dopamine synthesis capacity in responders (N=12) relative to patients with treatment-resistant illness (N=12). The most significant increase was in the head of the caudate nucleus (p=0.039, corrected at the family-wise error rate).
Tables
Media
References
References
1.
Lindenmayer JP: Treatment refractory schizophrenia. Psychiatr Q 2000; 71:373–384
2.
Howes OD, Kapur S: The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophr Bull 2009; 35:549–562
3.
Howes OD, Montgomery AJ, Asselin MC, Murray RM, Grasby PM, McGuire PK: Molecular imaging studies of the striatal dopaminergic system in psychosis and predictions for the prodromal phase of psychosis. Br J Psychiatry Suppl 2007; 51:s13–s18
4.
Howes OD, Montgomery AJ, Asselin MC, Murray RM, Valli I, Tabraham P, Bramon-Bosch E, Valmaggia L, Johns L, Broome M, McGuire PK, Grasby PM: Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry 2009; 66:13–20
5.
Howes OD, Bose SK, Turkheimer F, Valli I, Egerton A, Valmaggia LR, Murray RM, McGuire P: Dopamine synthesis capacity before onset of psychosis: a prospective [18F]-DOPA PET imaging study. Am J Psychiatry 2011; 168:1311–1317
6.
Howes O, Bose S, Turkheimer F, Valli I, Egerton A, Stahl D, Valmaggia L, Allen P, Murray R, McGuire P: Progressive increase in striatal dopamine synthesis capacity as patients develop psychosis: a PET study. Mol Psychiatry 2011; 16:885–886
7.
Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann JJ, Van Heertum RL, Gorman JM, Laruelle M: Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci USA 2000; 97:8104–8109
8.
Laruelle M, Abi-Dargham A, van Dyck CH, Gil R, D’Souza CD, Erdos J, McCance E, Rosenblatt W, Fingado C, Zoghbi SS, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB: Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci USA 1996; 93:9235–9240
9.
Kapur S, Zipursky R, Jones C, Remington G, Houle S: Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry 2000; 157:514–520
10.
Talbot PS, Laruelle M: The role of in vivo molecular imaging with PET and SPECT in the elucidation of psychiatric drug action and new drug development. Eur Neuropsychopharmacol 2002; 12:503–511
11.
Creese I, Burt DR, Snyder SH: Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 1976; 192:481–483
12.
Kapur S, Zipursky R, Jones C, Shammi CS, Remington G, Seeman P: A positron emission tomography study of quetiapine in schizophrenia: a preliminary finding of an antipsychotic effect with only transiently high dopamine D2 receptor occupancy. Arch Gen Psychiatry 2000; 57:553–559
13.
Abi-Dargham A, Laruelle M: Mechanisms of action of second generation antipsychotic drugs in schizophrenia: insights from brain imaging studies. Eur Psychiatry 2005; 20:15–27
14.
Wolkin A, Barouche F, Wolf AP, Rotrosen J, Fowler JS, Shiue CY, Cooper TB, Brodie JD: Dopamine blockade and clinical response: evidence for two biological subgroups of schizophrenia. Am J Psychiatry 1989; 146:905–908
15.
Duncan E, Wolkin A, Angrist B, Sanfilipo M, Wieland S, Cooper TB, Rotrosen J: Plasma homovanillic acid in neuroleptic responsive and nonresponsive schizophrenics. Biol Psychiatry 1993; 34:523–528
16.
McGuffin P, Farmer A, Harvey I: A polydiagnostic application of operational criteria in studies of psychotic illness: development and reliability of the OPCRIT system. Arch Gen Psychiatry 1991; 48:764–770
17.
Conley RR, Kelly DL: Management of treatment resistance in schizophrenia. Biol Psychiatry 2001; 50:898–911
18.
Kay SR, Fiszbein A, Opler LA: The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull 1987; 13:261–276
19.
Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR: What does the PANSS mean? Schizophr Res 2005; 79:231–238
20.
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 4th ed Washington, DC, American Psychiatric Association, 1994
21.
Andreasen NC, Carpenter WT, Kane JM, Lasser RA, Marder SR, Weinberger DR: Remission in schizophrenia: proposed criteria and rationale for consensus. Am J Psychiatry 2005; 162:441–449
22.
Martinez D, Slifstein M, Broft A, Mawlawi O, Hwang DR, Huang Y, Cooper T, Kegeles L, Zarahn E, Abi-Dargham A, Haber SN, Laruelle M: Imaging human mesolimbic dopamine transmission with positron emission tomography, part II: amphetamine-induced dopamine release in the functional subdivisions of the striatum. J Cereb Blood Flow Metab 2003; 23:285–300
23.
Patlak CS, Blasberg RG, Fenstermacher JD: Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab 1983; 3:1–7
24.
Carlsson A, Lindqvist M: Effect of chlorpromazine or haloperidol on formation of 3-methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol (Copenh) 1963; 20:140–144
25.
Miyamoto S, Duncan GE, Marx CE, Lieberman JA: Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry 2005; 10:79–104
26.
Der-Ghazarian T, Charntikov S, Varela FA, Crawford CA, McDougall SA: Effects of repeated and acute aripiprazole or haloperidol treatment on dopamine synthesis in the dorsal striatum of young rats: comparison to adult rats. J Neural Transm 2010; 117:573–583
27.
McGowan S, Lawrence AD, Sales T, Quested D, Grasby P: Presynaptic dopaminergic dysfunction in schizophrenia: a positron emission tomographic [18F]fluorodopa study. Arch Gen Psychiatry 2004; 61:134–142
28.
Samaha AN, Seeman P, Stewart J, Rajabi H, Kapur S: “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci 2007; 27:2979–2986
29.
Ginovart N, Wilson AA, Hussey D, Houle S, Kapur S: D2-receptor upregulation is dependent upon temporal course of D2-occupancy: a longitudinal [11C]-raclopride PET study in cats. Neuropsychopharmacology 2009; 34:662–671
30.
Kolakowska T, Williams AO, Ardern M, Reveley MA, Jambor K, Gelder MG, Mandelbrote BM: Schizophrenia with good and poor outcome, I: early clinical features, response to neuroleptics, and signs of organic dysfunction. Br J Psychiatry 1985; 146:229–239
31.
Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, Hwang DR, Huang Y, Haber SN, Laruelle M: Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry 2010; 67:231–239
32.
Reith J, Benkelfat C, Sherwin A, Yasuhara Y, Kuwabara H, Andermann F, Bachneff S, Cumming P, Diksic M, Dyve SE, Etienne P, Evans AC, Lal S, Shevell M, Savard G, Wong DF, Chouinard G, Gjedde A: Elevated dopa decarboxylase activity in living brain of patients with psychosis. Proc Natl Acad Sci USA 1994; 91:11651–11654
33.
Shotbolt P, Stokes PR, Owens SF, Toulopoulou T, Picchioni MM, Bose SK, Murray RM, Howes OD: Striatal dopamine synthesis capacity in twins discordant for schizophrenia. Psychol Med 2011; 41:2331–2338
34.
Elkashef AM, Doudet D, Bryant T, Cohen RM, Li SH, Wyatt RJ: 6-(18)F-DOPA PET study in patients with schizophrenia: positron emission tomography. Psychiatry Res 2000; 100:1–11
35.
Vernaleken I, Kumakura Y, Cumming P, Buchholz HG, Siessmeier T, Stoeter P, Müller MJ, Bartenstein P, Gründer G: Modulation of [18F]fluorodopa (FDOPA) kinetics in the brain of healthy volunteers after acute haloperidol challenge. Neuroimage 2006; 30:1332–1339
36.
Ito H, Takano H, Takahashi H, Arakawa R, Miyoshi M, Kodaka F, Okumura M, Otsuka T, Suhara T: Effects of the antipsychotic risperidone on dopamine synthesis in human brain measured by positron emission tomography with L-[beta-11C]DOPA: a stabilizing effect for dopaminergic neurotransmission? J Neurosci 2009; 29:13730–13734
37.
Gründer G, Vernaleken I, Müller MJ, Davids E, Heydari N, Buchholz HG, Bartenstein P, Munk OL, Stoeter P, Wong DF, Gjedde A, Cumming P: Subchronic haloperidol downregulates dopamine synthesis capacity in the brain of schizophrenic patients in vivo. Neuropsychopharmacology 2003; 28:787–794
38.
Grace AA, Bunney BS, Moore H, Todd CL: Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci 1997; 20:31–37
39.
Valenti O, Cifelli P, Gill KM, Grace AA: Antipsychotic drugs rapidly induce dopamine neuron depolarization block in a developmental rat model of schizophrenia. J Neurosci 2011; 31:12330–12338
40.
Carlsson A, Waters N, Holm-Waters S, Tedroff J, Nilsson M, Carlsson ML: Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol 2001; 41:237–260