Prefrontal cortex-related cognitive impairments in schizophrenia have been linked to disturbances in the inhibitory system, such as deficits in the GABA synthesizing enzyme glutamate decarboxylase (GAD67) (
1–
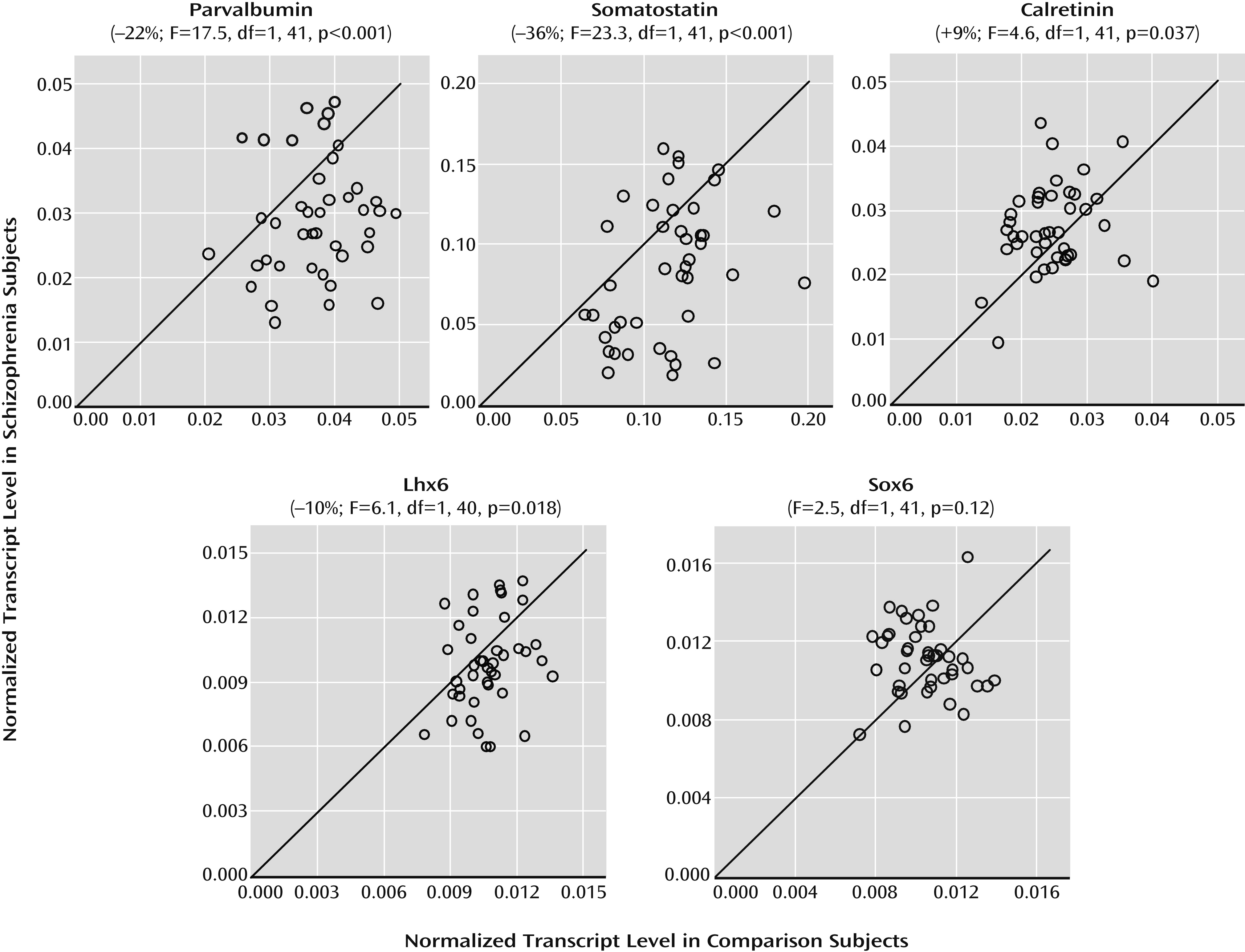
6). Alterations in cortical GABA neurons are most prominent in the subsets that contain the calcium-binding protein parvalbumin or the neuropeptide somatostatin but not in the subset that expresses the calcium-binding protein calretinin. For example, mean levels of parvalbumin, somatostatin, and GAD67 mRNAs in the prefrontal cortex have been consistently reported to be lower in cohorts of schizophrenia subjects, but not all schizophrenia subjects in each cohort had lower levels than their matched comparison subjects (
5,
7–
10). The pathogenetic mechanisms that lead to the molecular pathology of specific GABA cell types that are prominent in a subset of schizophrenia subjects are not known; however, factors related to the developmental origin of the different GABA neuron subpopulations may play a role.
Indeed, the specification of cortical GABA neurons into distinct subpopulations is related to the location of their origination and is regulated by cell type-specific transcription factors. For example, parvalbumin and somatostatin neurons originate from the medial ganglionic eminence of the subpallium in humans and rodents (
11–
16), whereas calretinin neurons derive from the subventricular zone of the dorsal pallium, at least in primates and humans (
14–
18). Furthermore, during the prenatal period, certain transcription factors (e.g., Lhx6 and Sox6) selectively regulate the ontogeny (i.e., cell type specification, tangential migration, and maturation) of parvalbumin and somatostatin neurons, but not calretinin neurons (
19–
22). A complete loss of Lhx6 or Sox6 in prenatal periods leads to slowed tangential migration to the cerebral cortex, impeded differentiation into parvalbumin and somatostatin neurons, and altered GAD67 levels (
19–
22). These lines of evidence suggest that in schizophrenia, early developmental disturbances, such as altered expression of cell type-specific transcription factors, may lead to persisting deficits that predominantly affect parvalbumin and somatostatin neurons, but not calretinin neurons.
Although it is not feasible to directly study embryonic cortical GABA neuron ontogeny in schizophrenia, some postmortem studies of adult schizophrenia subjects have provided evidence suggestive of arrested migration of somatostatin neurons (
23) and of failure of parvalbumin neurons to develop a GABA-ergic phenotype (
7). Interestingly, Lhx6 and Sox6 continue to be selectively and robustly expressed by virtually all parvalbumin and somatostatin neurons in adult cortex of humans and rodents (
13,
19,
21,
22,
24). In this study, we sought to determine whether mRNA levels for Lhx6 and Sox6 in postmortem human prefrontal cortex are deficient in schizophrenia and whether such deficits are especially prominent in schizophrenia subjects with clear deficits in parvalbumin, somatostatin, and GAD67 mRNAs.
Discussion
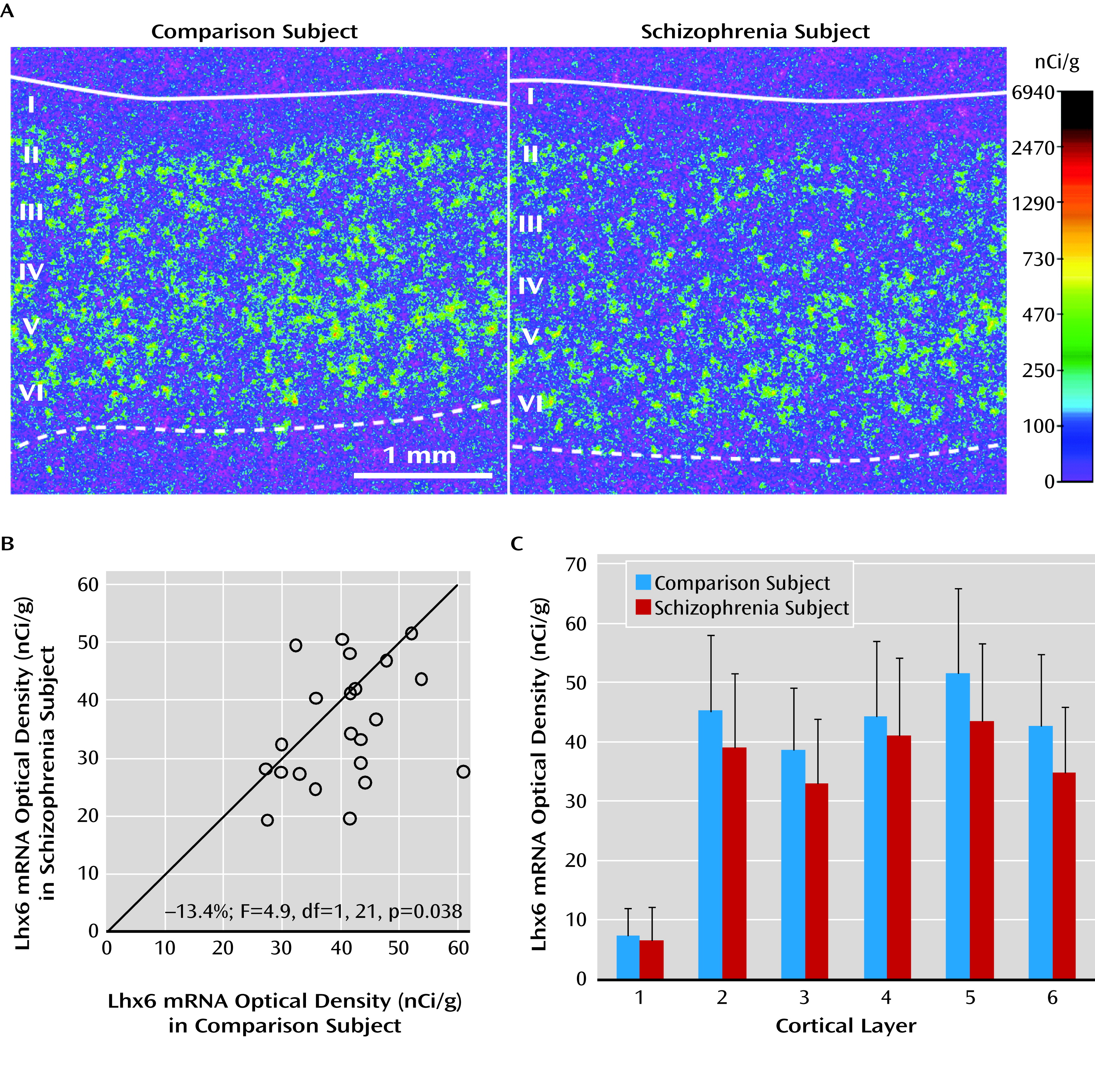
In the prefrontal cortex of schizophrenia subjects, we found deficits in mRNA levels for Lhx6, an ontogenetic transcription factor selectively expressed by parvalbumin and somatostatin neurons in humans (
24). Furthermore, our findings of lower mRNA levels for parvalbumin and somatostatin are remarkably similar to reports in other cohorts of schizophrenia subjects (
5,
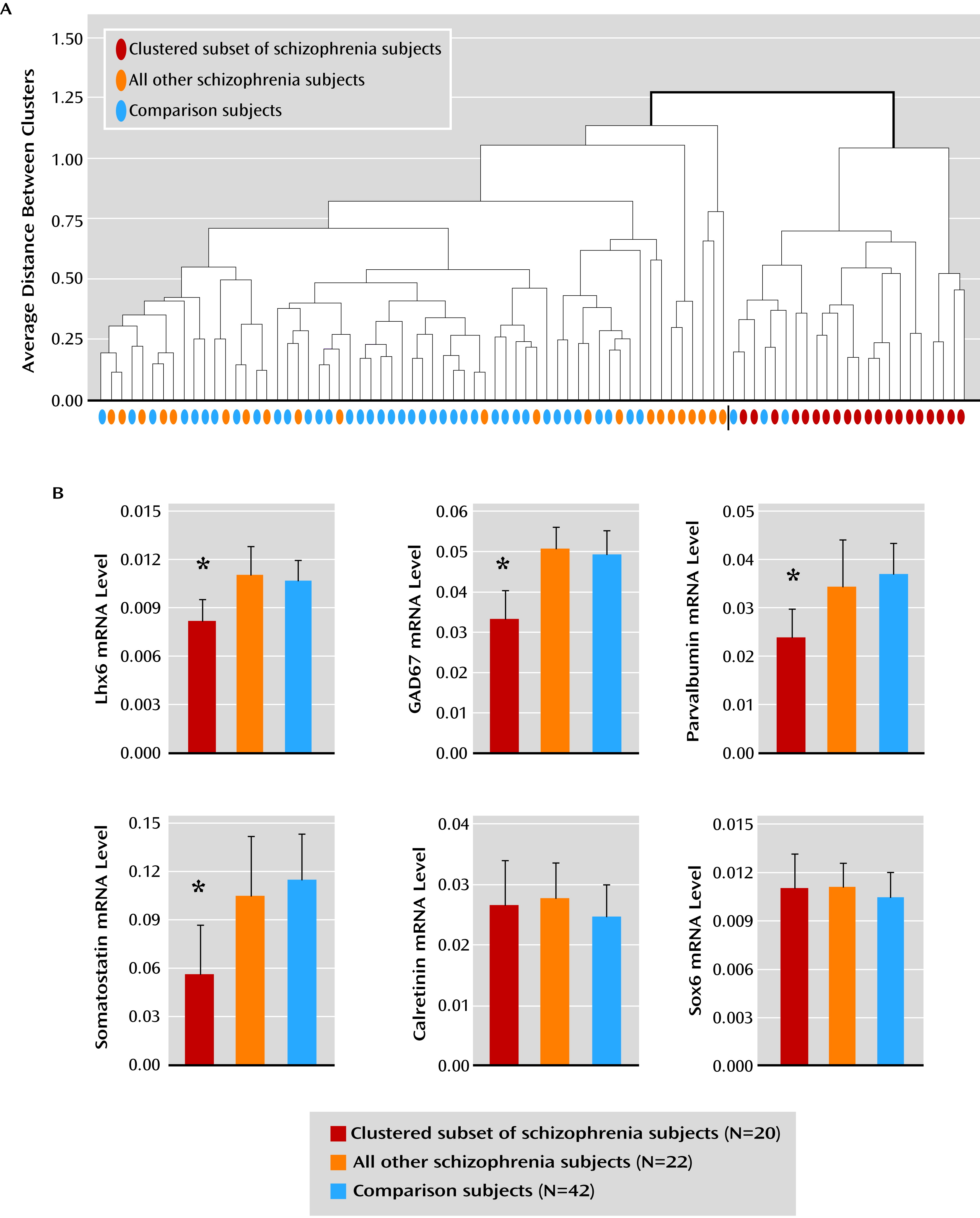
10). Interestingly, a subset of schizophrenia subjects was identified by the presence of consistent deficits in Lhx6, GAD67, parvalbumin, and somatostatin mRNAs. In contrast, not all GABA neuron markers were lower in this subset of schizophrenia subjects (e.g., calretinin), and not all mRNA species localized to parvalbumin and somatostatin neurons were deficient (e.g., Sox6). Taken together, these data suggest that deficient Lhx6 mRNA expression may contribute to alterations in phenotypic markers in prefrontal parvalbumin and somatostatin neurons in a subset of schizophrenia subjects.
Lhx6 is a transcription factor expressed by (future) parvalbumin and somatostatin neurons as they migrate from the medial ganglionic eminence to the cerebral cortex (
11–
14,
19,
20) as early as 7 weeks’ gestation in humans (
16), and Lhx6 continues to be strongly expressed in this cell type-specific manner in adult human cortex (
24). The pathogenetic consequences of low Lhx6 levels may depend on the developmental stage at which Lhx6 deficits first emerge in the disorder. For example, deficits in Lhx6 at the earliest gestational periods impair the migration of cortical parvalbumin and somatostatin neurons (
19,
20). In schizophrenia, the densities of cortical Lhx6-positive neurons and somatostatin-positive neurons (
9) are lower in the gray matter while the density of somatostatin-positive neurons is higher in the interstitial white matter (
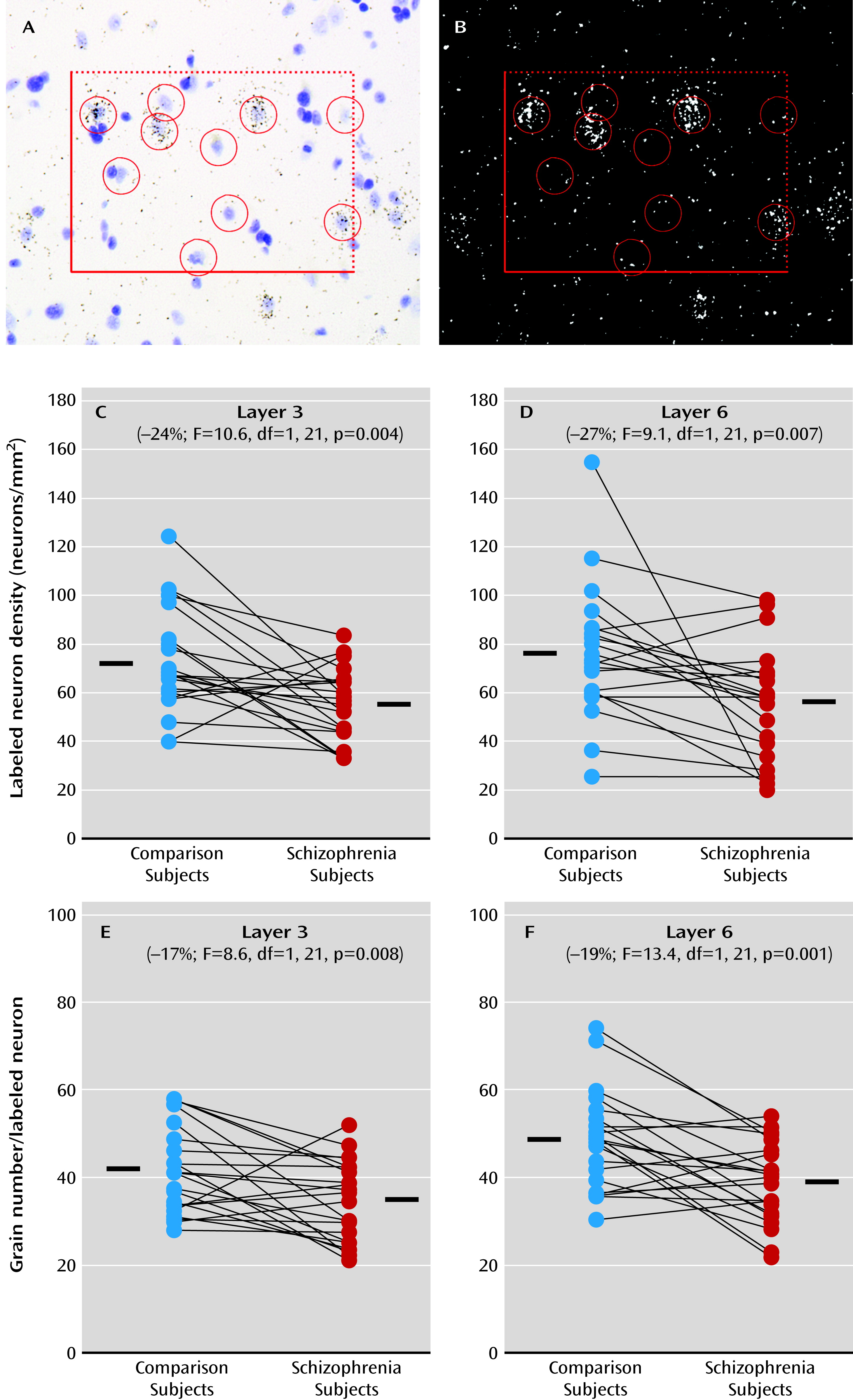
23); together, these findings are suggestive of arrested migration of some somatostatin neurons. Alternatively, a loss of Lhx6 that occurs after migration might interfere with the development of the GABA-ergic phenotype. For example, a lower density of cortical Lhx6-positive neurons could mean that these neurons are still present but have undetectable levels of Lhx6 mRNA. Consistent with this interpretation, the density of prefrontal parvalbumin mRNA-containing neurons was not altered in schizophrenia, but approximately 50% of these neurons lacked detectable GAD67 mRNA (
7). As a third alternative, deficits in Lhx6 may appear in late adolescence/early adulthood after cellular maturation has been completed. In this case, the clustering of Lhx6, GAD67, parvalbumin, and somatostatin mRNA deficits in a subset of schizophrenia subjects may instead reflect the shared downstream consequences of some other upstream pathogenetic event that selectively affects parvalbumin and somatostatin neurons. However, our failure to detect a change in Sox6 mRNA, which is selectively expressed by cortical parvalbumin and somatostatin neurons (
21,
22), is inconsistent with a general disease effect that affects all transcripts localized to these neurons. Finally, the absence of an effect of age on Lhx6 mRNA levels in schizophrenia suggests that lower Lhx6 mRNA levels are not attributable to a prolonged neurodegenerative process. Since our study was designed to detect alterations present in adulthood, we cannot directly determine the developmental stage at which deficits in Lhx6 mRNA levels, or any other GABA neuron markers, occur in the disorder. Interestingly, though, cognitive developmental delays have been reported in the premorbid stages of schizophrenia (
32), even before age 1 (
33). Thus, these lines of evidence suggest that the pattern of molecular abnormalities in prefrontal GABA neurons seen in a subset of schizophrenia subjects may reflect the long-lasting consequence of disturbances in the ontogeny of parvalbumin and somatostatin neurons that might have begun early in prenatal life.
The finding that deficits in certain GABA neuron-related mRNAs cluster together in a subset of schizophrenia subjects may help inform novel diagnostic strategies and individualized treatment approaches in schizophrenia. This subset of schizophrenia subjects with clear GABA neuron-related deficits may not necessarily have a more severe form of the illness, as suggested by the lack of difference in markers of illness severity from other schizophrenia subjects. However, one may predict that schizophrenia-related deficits in gamma band oscillations (
34,
35), which are subserved by cortical parvalbumin neurons (
36) and are important for prefrontal cortex-related cognitive processes (
37), may be greater in the low-GABA-marker subset of schizophrenia subjects (
38). Furthermore, approaches to clinically identifying schizophrenia patients with GABA neuron-related deficits might include positron emission tomography studies of shifts in extracellular GABA levels (
39) or magnetic resonance spectroscopy (MRS) studies to quantify total cortical GABA levels. Interestingly, the evidence for two subsets of schizophrenia subjects, with or without robustly lower cortical GAD67 mRNA levels (
Figure 4), suggests that inconsistencies in reports of cortical GABA levels by MRS (
40–
42) may reflect differences in the sampling of schizophrenia subjects (i.e., different studies may include a variable proportion of schizophrenia subjects with and without deficits in GABA neuron-related mRNAs). In addition, allelic variants or altered histone methylation of the promoter regions of GABA neuron-related genes, such as GAD1 (
4,
43), that affect gene expression and have been associated with an increased risk for schizophrenia may be even more common in the low-GABA-marker subset of schizophrenia subjects. Finally, personalized medicine approaches, such as using GABA agents (
44,
45) in schizophrenia patients with identified cortical GABA deficits, may increase the likelihood of a beneficial effect on cognitive functioning.