Epidemiological studies have documented high rates of comorbidity among alcohol, nicotine, and illicit drug dependence disorders (
1–
5). Comorbidity suggests that these nosologically distinct disorders are caused, in part, by common etiological processes (
6–
8). In substance use disorders, the etiology shared among disorders indexed by comorbidity has been referred to as “externalizing” or “disinhibitory psychopathology” (
9).
While comorbidity among substance use disorders has been extensively studied cross-sectionally—typically for lifetime prevalence rates—few studies have examined how comorbidity changes over time, an important topic given the large changes in incidence over the lifespan. Specifically, substance use tends to emerge in middle adolescence, increases substantially throughout adolescence, peaks in the early twenties, plateaus, and then decreases in the late twenties (
10). Whether the rates of comorbidity among substance use disorders remain consistently high, however, is unknown. For example, rates of comorbidity might decline, suggesting that individuals begin to specialize in their substance use over time.
To investigate comorbidity across time, we used a large longitudinal twin study to examine changes in correlations among substance use disorder symptom counts over time. Assessments coincided with key developmental transitions in substance use, including before initiation (age 11), initiation (age 14), regular use (age 17), heavy use and dependence (ages 20 and 24), and the period when individuals decrease their use or exhibit patterns of persistent substance use problems (age 29). First, we examined patterns of mean-level change in nicotine, alcohol, and marijuana dependence symptoms. Second, we fitted a single common factor model to account for the correlations among incident symptom counts of nicotine, alcohol, and marijuana dependence at each age. This common factor model then allowed us to test for changes in the contributions of common and specific etiological influences on these symptom counts across time. Third, we used standard twin models to estimate genetic and environmental influences on the common factor over time.
In addition to analyzing the full sample, we also fitted separate models to a subsample of early-onset users. We did so because estimates of correlations at earlier ages may be affected by a minority of high-risk individuals who tend to exhibit dependence symptoms for multiple substances. Within this high-risk sample, correlations among dependence symptoms may remain high into adulthood. This could be obscured, however, in analyses of the full sample as substance use becomes more normative in adulthood. That is, if the correlations among substance use disorders are lower for the larger group of later-onset users, what is actually an artifact of early- instead of later-onset use would appear to be an overall decline in the correlations among substance use disorders. We thus performed our analyses both for the full sample and in a subsample of participants who had at least one symptom by their assessment at age 17, earlier than the age at which use of any of these substances becomes legal in the United States.
Method
Sample
Participants (N=3,762; 52% female) were drawn from the Minnesota Twin Family Study, a community-representative longitudinal study of Minnesota families (
11). The younger twin cohort (N=2,510; 51% female) was first assessed at age 11 during the years 1988–2005. The older cohort (N=1,252; 54% female) was first assessed at age 17 during the years 1989–1996. Members of the age 11 cohort were invited to participate in follow-up assessments at ages 14 and 17, and all twins were invited to participate in follow-up assessments at ages 20, 24, and 29. Cohorts were combined for all analyses. Participants received modest payments for their assessments. Written assent or consent was obtained from all participants, including the parents of minor children, and all study protocols were approved by the University of Minnesota institutional review board.
Additional analyses were conducted with a subsample of participants who had at least one symptom by their assessment at age 17 for nicotine, alcohol, or marijuana dependence. This resulted in an early-use subsample of 580 male and 486 female participants.
Pooling across cohorts in the full sample, the actual mean ages at assessment were 11.8 (SD=0.4), 14.9 (SD=0.6), 17.8 (SD=0.7), 21.1 (SD=0.8), 25.0 (SD=0.9), and 29.5 (SD=0.7) years. Participation rates ranged from 87.3% to 93.6% for the follow-up assessments. To examine attrition, we compared the results from 17-year-olds who completed the adult assessments at ages 20, 24, and 29 against those who did not. For male participants, the Cohen’s d values for mean differences in dependence symptoms at age 17 between those who completed the later assessments (N=1,570) and those who did not (N=238) were 0.00, −0.08, and 0.09 for nicotine, alcohol, and marijuana, respectively. For female participants, the Cohen’s d values for similar comparisons were −0.19, −0.01, and 0.13 (p>0.05 in all cases).
Measures
Diagnostic symptom counts were obtained during in-person interviews with trained interviewers. In a consensus process, graduate students and staff with advanced training in clinical assessment reviewed cases to verify the presence of symptoms.
Assessments at ages 11 and 14 used the Diagnostic Interview for Children and Adolescents (
12) to examine DSM-III-R nicotine dependence, alcohol dependence or abuse, and marijuana dependence or abuse. Assessments at later ages used a modified version of the Substance Abuse Module (
13) of the Composite International Diagnostic Interview (CIDI [
14]) to assess DSM-III-R symptoms of substance use disorders. Abuse and dependence symptoms were collapsed for alcohol and marijuana. We also obtained mothers’ reports of their children’s symptoms at ages 11, 14, and 17. The follow-up assessments at each age covered the interval that had elapsed since the last assessment. We used a best-estimate approach (
15) whereby a symptom was considered present if reported by either the child or the mother. Diagnostic interrater reliability of substance use disorders was greater than 0.91 (
16). To rule out possible informant effects, all analyses were repeated using only the child as the informant, and the pattern of results was identical.
Analysis of Change in Comorbidity
Bivariate correlations were computed using Spearman’s rank-order correlation statistic, which is robust to departures from bivariate normality (
17). We used confirmatory factor analysis (
18) to model the pattern of correlations among substance use disorders over time. For each assessment age, a single factor was fitted to account for correlations among symptom counts of alcohol, nicotine, and marijuana dependence. Because of prohibitively low variance, symptoms at age 11 were not included in the model. This resulted in a model with five general factors, one for each age of assessment and each representing the covariance among substance use disorders at each age. All factors were allowed to correlate, and all same-drug residuals were allowed to correlate across ages to account for the within-person correlated nature of the longitudinal data. Loadings were standardized. Thus, the variance of the symptom count variables can be modeled as a function of the general and residual (substance-specific) factors:
where “var” denotes variance. Since the variance of the general factor was set to 1, the variance in a standardized symptom count accounted for by the general factor is simply the square of the factor loading. To obtain a single estimate of comorbidity for each age of assessment (i.e., a single estimate of the variance in the three symptom counts accounted for by the general factor), we calculated the mean squared factor loadings at each age. The mean squared loading provides a reasonable metric to test for changes in correlations because it is directly proportional to the magnitude of correlations among the symptom count variables (i.e., higher correlations among the symptom counts result in greater mean variance in the symptom counts accounted for by the general factor). To test for significant differences, we constrained the mean squared loading to be the same across assessment waves, and we conducted likelihood ratio tests to evaluate the change in model fit according to standard practice (
19).
In addition, because the sample was composed of twins, we used standard twin modeling to decompose the general factor variances into three components: additive genetic (A), shared environmental (C), and nonshared environmental (E) components. All analyses were conducted in the same model rather than separate phenotypic and biometric models. Because we set the variance of the general factor to 1, the sum of the A, C, and E variance components of the general factor is also 1. To obtain the average variance in the symptom counts accounted for by genetic influences on the factor, one simply substitutes the term “var(general factor)” in the above equation with the corresponding genetic, shared environmental, or nonshared environmental component of the general factor variance. For example, a mean squared loading of 0.4 at age 17 would indicate that 40% of the variance in the symptom counts was due to the general factor. If the additive genetic component A of the age 17 factor was 0.7, then the genetic influence of the general factor onto the symptom counts would be 0.4×0.7=0.28. That is, 28% of the symptom count variance would be due to the genetic influence of the factor.
Investigating the biometric decomposition of symptom count correlations is only applicable to the full sample because the subsample analysis of individuals symptomatic by age 17 was, by definition, a within-individual analysis and disregards unaffected or later-affected (after age 17) co-twins. Resulting biometric decompositions would be difficult to interpret because only those pairs of twins who were symptomatic by age 17 were included in the cross-twin correlations.
Analyses were conducted using R, version 2.10.1 (
20), and the factor analysis was conducted using OpenMx, version 1.0.6 (
21). Missing data was handled using full information maximum likelihood. We evaluated model fit using chi-square tests and the difference in the Akaike information criterion (
22) between the saturated and alternative models (positive values indicate that the alternative provides better fit) separately for the four samples. We also report the root mean square error of approximation, where values of 0.06 or less indicate a very good fit (
23). The chi-square test provides an exact test of model fit, but it is sensitive to sample size and the magnitude of correlations among measures and is always significant (p<0.05) when statistical power is high (e.g., in large samples). It is often significant even when the model provides an accurate and useful representation of the data (
24). Therefore, other fit indices have been developed and are primarily used to evaluate model fit. The root mean square error of approximation attempts to correct deficiencies of the chi-square by adjusting for the degrees of freedom and sample size. Conventional root mean square error of approximation cutoffs are 0.08 for good fit and 0.05 for very good fit. The Akaike information criterion has strong theoretical properties in that the selected model is expected to fit best upon cross-validation (
22).
Results
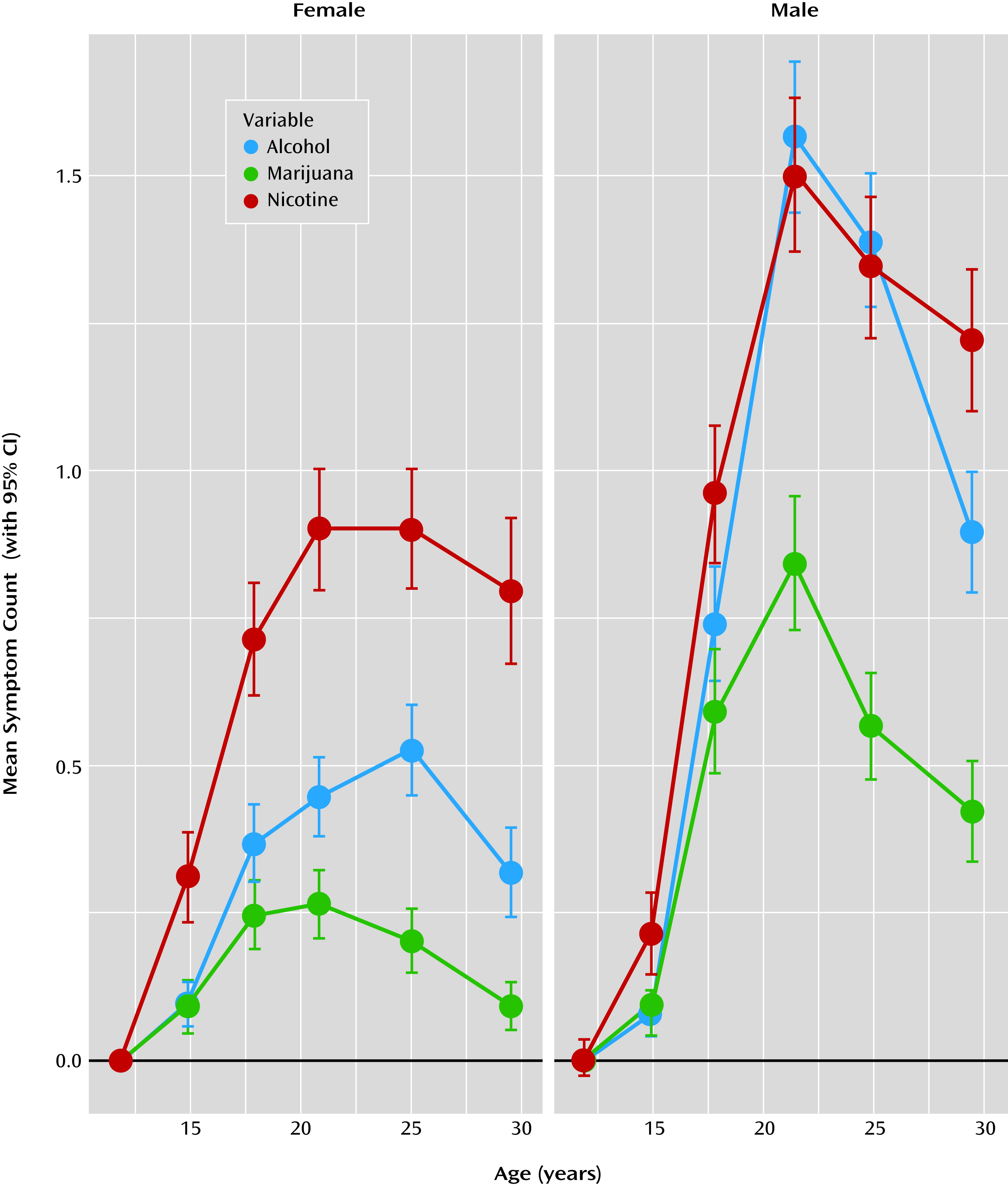
Longitudinal trends in symptom counts were somewhat different for male and female participants (
Figure 1 and
Table 1). For male participants, symptom counts significantly increased from age 11 to late adolescence, peaked during the early twenties, and declined thereafter. For female participants, a more prolonged plateau during the late teens and early twenties was observed, with a marked drop only by age 29. Mean symptom counts increased more rapidly for male than for female participants, with teenage boys maintaining higher mean-level symptoms after age 14. Variances also increased during adolescence and decreased during the late twenties.
The prevalence rates for each disorder and the proportion of individuals who had ever used each substance for each age are reported in
Table 1. The mean age at initiation was 14.4 years (SD=3.4) for nicotine, 15.5 years (SD=2.6) for alcohol use without parental permission, and 16.5 years (SD=2.5) for marijuana. The mean age at symptom onset was 17.5 years (SD=2.7) for nicotine, 18.2 years (SD=4.4) for alcohol, and 17.2 years (SD=2.3) for marijuana.
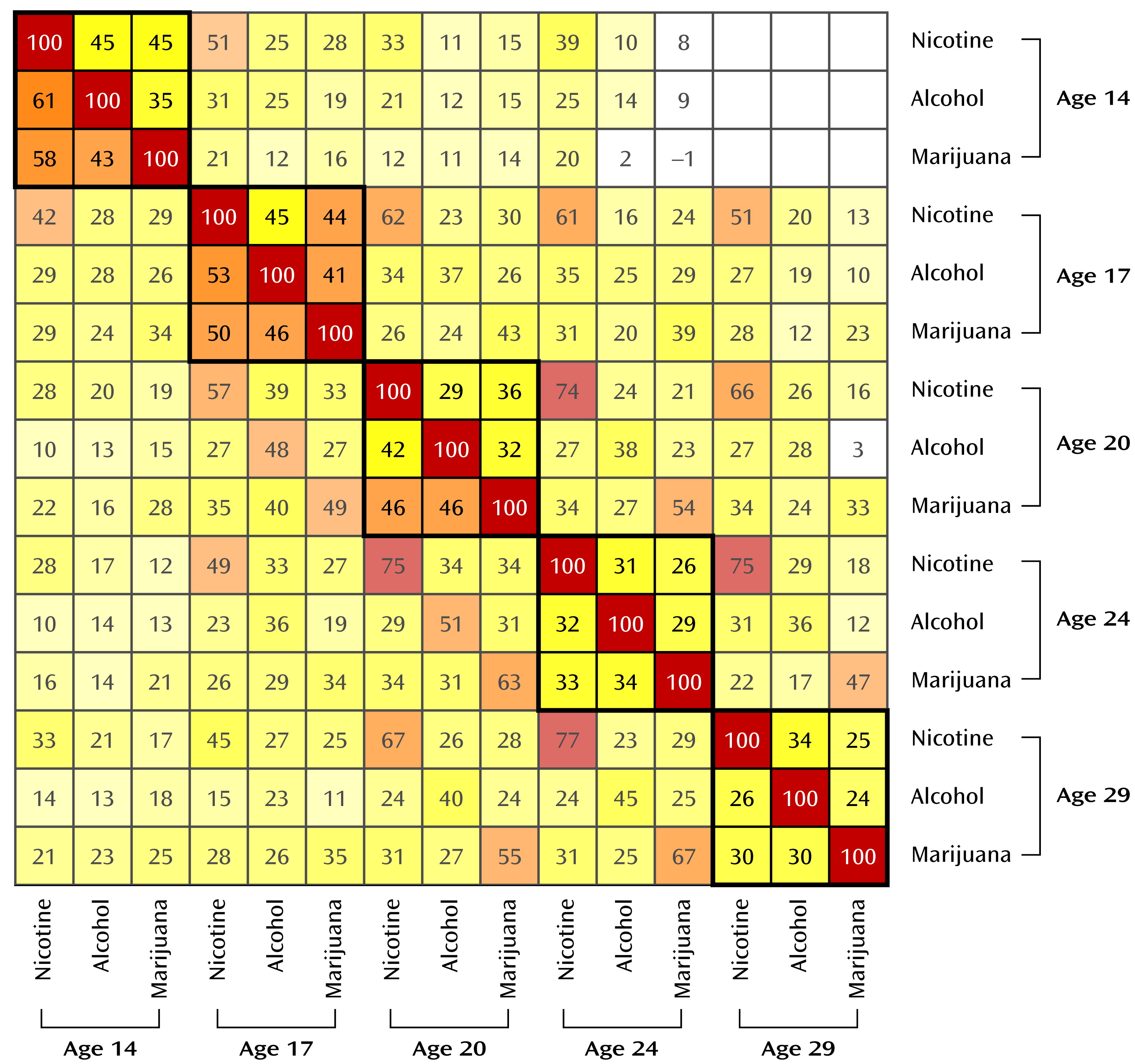
Cross-drug correlations decreased with age, as can be seen in the full bivariate correlation matrix in
Figure 2. For example, for male participants, the correlation between nicotine and alcohol declined from 0.61 at age 14 to 0.26 at age 29.
Factor models were fitted to provide a formal test of changes in the cross-drug correlations. The model fit in the full male sample was very good (χ2=216.66, df=170, p=0.009; ΔAkaike information criterion=123.33; root mean square error of approximation=0.02). Model fit in the full female sample was also good (χ2=432.32, df=170, p<0.001; ΔAkaike information criterion=−92.32; root mean square error of approximation=0.05). The model fit was good in both the male (χ2=137.45, df=170, p=0.97; ΔAkaike information criterion=202.55; root mean square error of approximation<0.01) and female (χ2=133.42, df=106, p=0.04; ΔAkaike information criterion=78.6; root mean square error of approximation=0.04) early-use subsamples.
For each model, the size of the standardized factor loadings generally decreased with age, declining from 0.6–0.8 at age 14 to 0.3–0.6 at age 29, as reported in
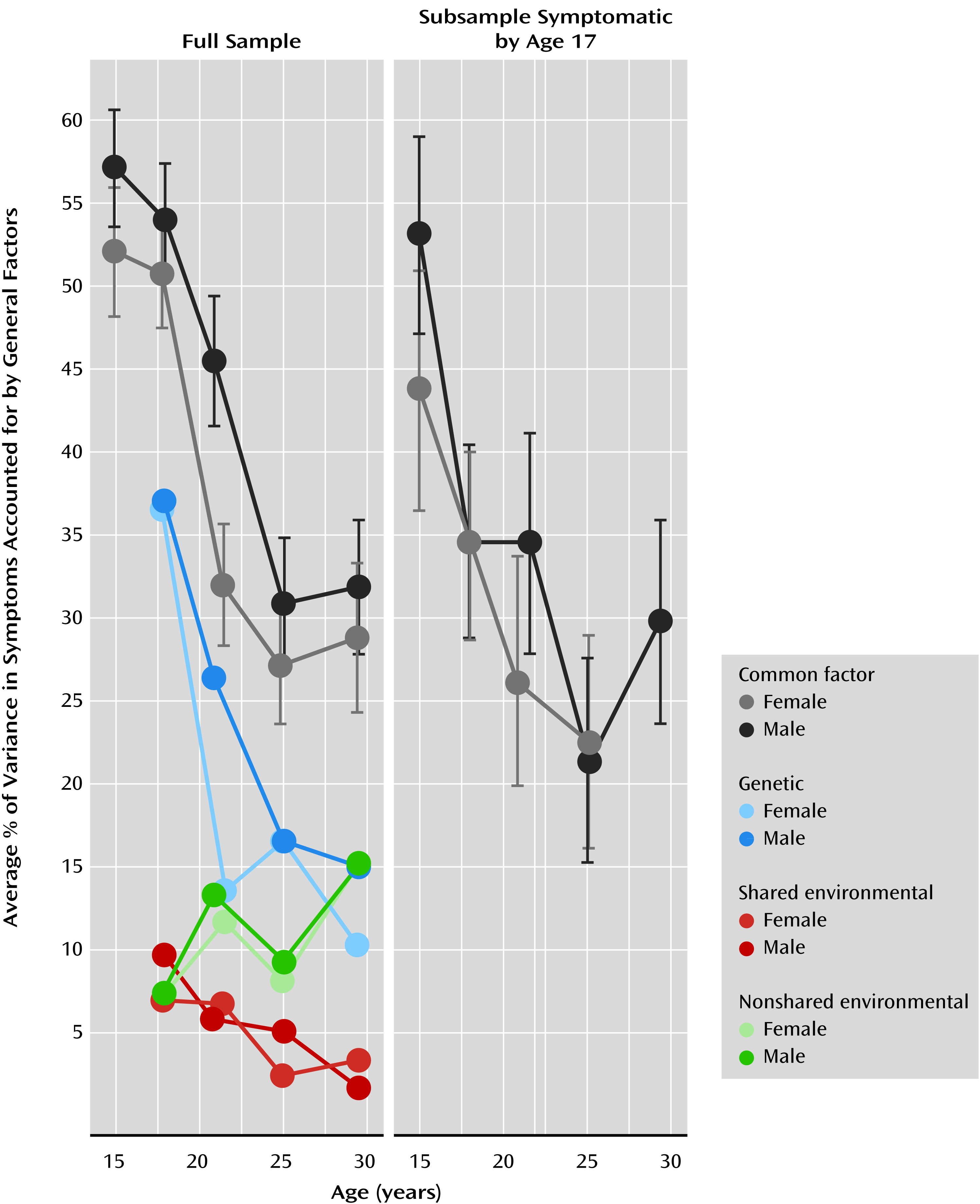
Table 2. The table also shows (as do the gray lines in
Figure 3) that the mean squared loading, which served as our metric of change, decreased with age. Recall that the mean squared factor loading provides the average variance in the symptoms accounted for by the general factor. To illustrate how these estimates are calculated, we entered the loadings from
Table 2 into equation 1 for the full female sample. The mean squared loading was (0.65
2+0.71
2+0.79
2)/3=0.52 at age 14 and (0.69
2+0.53
2+0.33
2)/3=0.29 at age 29. To test whether the decrease from age 14 to age 29 was statistically significant, we constrained the mean squared loadings to be same at each age and examined decrement in model fit. The likelihood ratio test indicated a significant decrease in the mean squared loading from age 14 to age 29 for all samples: full male sample (χ
2=158.4, df=4, p=3.2×10
−33), full female sample (χ
2=146.97, df=4, p=9.1×10
−31), male subsample of early users (χ
2=54.66, df=4, p=3.8×10
−11), and from age 14 to age 24 for the female subsample of early users (χ
2=21.36, df=3, p=8.9×10
−5). We tested for sex differences by comparing the fit of an unconstrained model to that of a model that constrained the mean squared loading to be equal across male and female participants. Relative to young men, young women showed an earlier decrease in comorbidity at age 20 (χ
2=24.18, df=1, p=8.8×10
−7), but not at any other ages.
Table 2 lists the additive genetic, shared environmental, and nonshared environmental components of each age’s general factor for each sample. While these estimates are useful, they must be multiplied by their respective factor loadings to determine the genetic and environmental impact on the individual symptom counts. To do this, we replaced the var(factor) term in equation 1 with the A, C, and E variance components. The resulting values are displayed for each age in
Figure 3. Note that the scaling is such that adding the A, C, and E components reproduces the mean squared loading (the mean squared loading is also listed in
Table 2). For example, for men at age 29, the phenotypic variance accounted for by the general factor was 32%, which was composed of 15% (0.32×0.47×100%) additive genetic variance, 15% (0.32×0.48×100%) nonshared environmental variance, and 2% (0.32×0.05×100%) shared environmental variance. Unfortunately, symptoms were expressed relatively infrequently by both members of a female twin pair at age 14, rendering the age 14 additive genetic, shared environmental, and nonshared environmental components poorly estimated and hence excluded from
Figure 3 (along with the male estimates for consistency).
We tested for change in heritability, shared environmental, and nonshared environmental estimates (see
Figure 3) by fixing relevant parameters to be equal at ages 17, 20, 24, and 29, testing for decrements in model fit. For example, to test for a change in heritability, we fixed the A component multiplied by the mean squared loading of each general factor to be equal, and we tested the fit of this model against the original model where all components are freely estimated. The decrease in heritability was significant for male (χ
2=23.86, df=3, p=2.7×10
−5) and female (χ
2=26.6, df=3, p=7.0×10
−6) participants. Thus, it appears that the vast majority of the phenotypic decline is the result of a decline in genetic variance. The increase in nonshared environment was also significant for male (χ
2=15.73, df=3, p=0.001) and female (χ
2=11.48, df=3, p=0.009) participants. Changes in shared environment did not reach significance for either sex.
Discussion
We examined changes in the correlations among symptoms of nicotine, alcohol, and marijuana abuse and dependence from age 11 to age 29. As shown in
Figure 3, male and female participants experienced significant declines in these correlations from adolescence to adulthood. This was also true for an early-onset subsample of individuals with at least one symptom of nicotine, alcohol, or marijuana dependence by age 17. Correlation among disorders or their symptoms is evidence that those disorders share etiology. Our results suggest that the shared etiology contributing to nicotine, alcohol, and marijuana dependence symptoms diminish over time. That is, younger individuals tended to use these three substances indiscriminately, whereas older individuals began to show a preference for one substance over the others. Despite decreases in the correlations, the rates of use (
Figure 1) continued to climb throughout late adolescence and early adulthood. Finally, after age 17 the correlations among symptoms became less attributable to pleiotropic genetic effects and increasingly a result of nonshared environmental influences (
Figure 3), indicating that the types of etiological processes contributing to the variation in use of multiple drugs gradually changes during the transition to adulthood.
Several processes might contribute to the transition from general to specific influences. For one, adolescents are more impulsive and risk-taking than adults (
25). Personality traits such as disinhibition and sensation-seeking are not predispositions to the use of any particular substance but rather the use of whatever substances might be available (
9,
26). Additionally, neurodevelopmental changes relevant to behavioral disinhibition continue throughout adolescence. For example, by adolescence, the nucleus accumbens—important in reward sensitivity—is well developed but poorly regulated by a still maturing prefrontal cortex (
27,
28), resulting in deficits of top-down control over the reward system that slowly improve in early adulthood. This developmental window is the same time period in which we observed decreases in the comorbidity among different substance use disorders, suggesting that disinhibitory mechanisms with known neurological substrates may be in play. Further supporting this hypothesis is that the earlier decline in comorbidity for female relative to male participants from age 17 to age 20 (
Figure 3) is consistent with the earlier pubertal (
29), cortical (
30), and personality (
31) maturation in girls compared with boys.
Our results are consistent with individual differences in drug reinforcement. That is, initial drug use that begins in adolescence is characterized by relatively indiscriminate experimentation. Because of individual differences in the reinforcing effects of different drugs, however, people may eventually restrict their use to those drugs that provide the greatest reinforcement. There are myriad etiological mechanisms relevant to individual differences in drug reinforcement, including drug metabolism effects, drug availability, social rewards and punishments, and differences in drug-specific neurological sensitivity that may be further moderated by drug and alcohol neurotoxic mechanisms (
26,
32). Future research might consider these covariates in characterizing the transition to substance specialization.
While our results are consistent with the theory that disinhibition accounts for comorbidity among substance use disorders, they are not entirely inconsistent with the gateway hypothesis (
33). The gateway hypothesis holds that using one drug leads to using other drugs, perhaps as a result of the experienced high and a greater desire to obtain bigger and better highs. If correct, the theory would predict the opposite pattern of comorbidity we observed; correlations should be low at younger ages and increase over time. At younger ages, most people would not yet have used their first or second gateway drug, and they would not have had time to explore other drugs. Over time, the correlations among drugs should increase as the initial gateway drug use would cause them to use other drugs. In fact, we found that as drug dependence symptoms increased (
Figure 1), the magnitude of the associations among different drugs decreased (
Figure 3); this is opposite to the pattern predicted by the gateway theory. This conclusion is consistent with a growing body of literature showing results that are inconsistent with parts of the gateway theory (
1,
9,
34–
36). That said, the increase in nonshared environmental effects observed in our sample could contain etiology analogous to a gateway process (i.e., environments that contribute to multiple drug dependence in one person but not in that person’s twin). While the range of possible environments is vast and the effect is small, it could very well include drug use (e.g., impaired cognition caused by neurotoxicity) or other risk factors for nonspecific drug use, such as occupational, social, and legal problems.
Our study has some limitations. The use of a community-representative sample, while itself a strength, may not apply to clinical populations. The fact that our results were consistent for the subsample of individuals who were symptomatic at age 17 suggests this is not a major concern. We used symptom counts because symptoms are relevant to the DSM clinical literature and provide measurement consistency from ages 11 to 29. In supplementary analyses we evaluated other measures of quantity and frequency of nicotine, alcohol, and marijuana use (not shown). These measures had higher means and variances at younger ages, and analyses resulted in the same trends as presented here for symptoms.
In the United States, adolescent development is confounded with substance-use-relevant environmental changes. Adolescents gradually experience greater autonomy and financial freedom from caregivers. The purchase of tobacco and alcohol becomes legal at ages 18 and 21, respectively, while marijuana use is always illegal. Undoubtedly, these environmental influences affected symptom means and correlations. However, one cannot disentangle these influences from other behavioral and neurological changes, at least within a single culture. Cross-cultural and cross-generational studies (e.g., comparisons between societies that differ in drug laws) are required to unravel maturational and environmental changes during development.