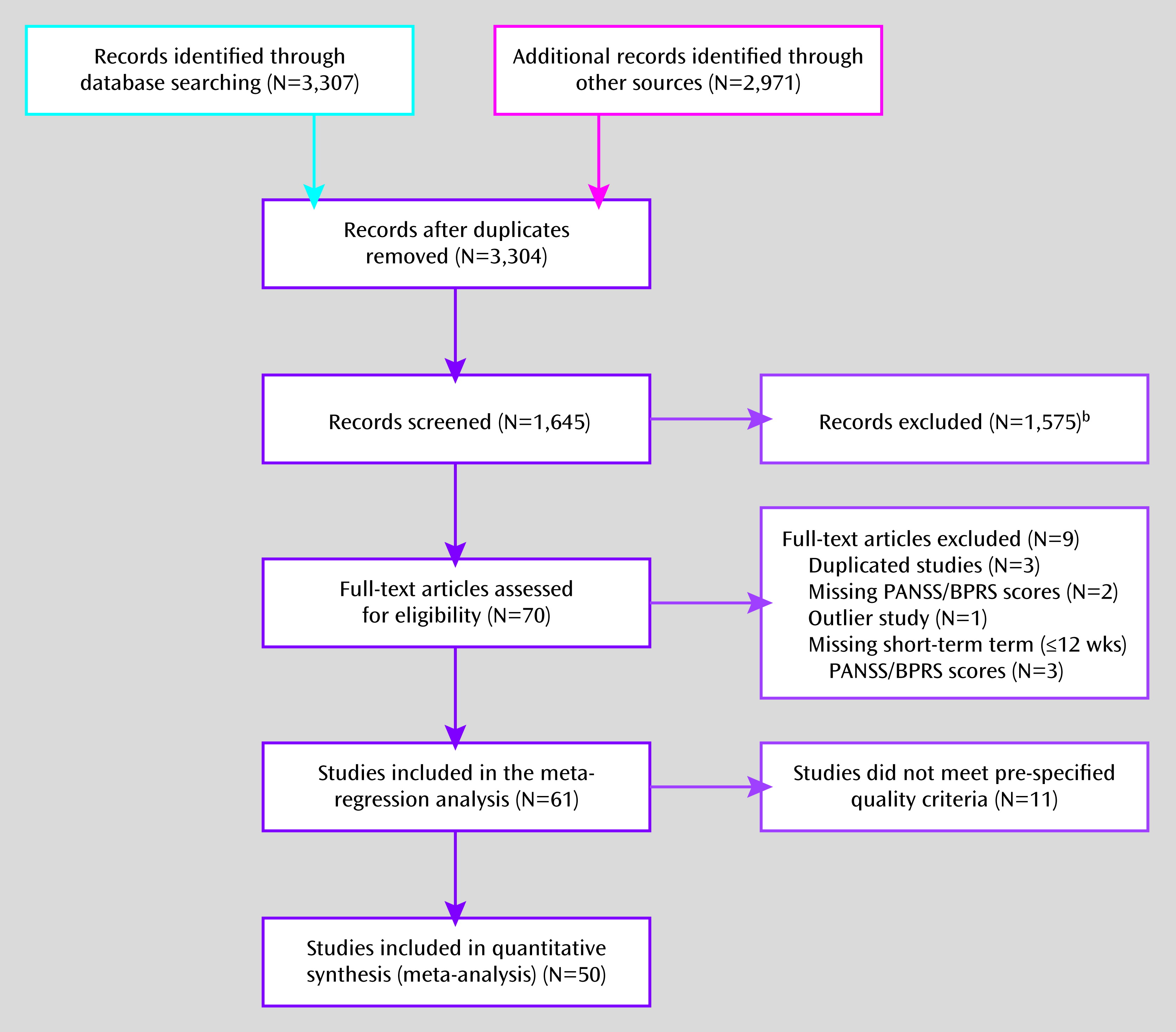
The inclusion of placebo control groups has been standard practice, and often a regulatory requirement, in randomized psychopharmacology trials despite some ethical concerns (
1–
3). An increasing placebo response in recent antipsychotic trials presents a major challenge in psychopharmacologic drug development (
4–
6), with studies reporting a trend toward diminished drug-placebo differences over time (
4,
6,
7). Reported placebo responses have ranged from 25% to 75% in psychopharmacology trials (
8), a trend contributing to numerous trial failures. In psychiatric trials, these failures often become apparent only late in the development process and can thus be particularly costly because of the intrinsically longer investigational path required for psychiatric agents (
9,
10).
There is a paucity of empirical data addressing placebo response in schizophrenia (
4–
7,
11,
12), and systematic analyses to date have been inconclusive. A meta-analysis of 32 randomized controlled trials conducted between 1970 and 2000 (
13) reported substantial variation in placebo effect size and identified trial duration as the only significant moderator affecting the reported placebo effect sizes. Another meta-analysis of 27 antipsychotic trials conducted between 1997 and 2008 (
14) found that increased placebo response was associated with a decrease in the percentage of patients assigned to receive placebo, a larger number of country sites involved in the study, and a greater percentage of female subjects involved, but not by other factors examined (year the trial was started, number of treatment arms, number of sites and regions, mean baseline symptom severity, trial duration, and dosing regimen). Systematic reviews of drug treatment effect sizes in schizophrenia trials have also been conducted, and they have found only small to modest effect sizes (
7,
11,
14,
15). A better understanding of factors underlying rising placebo responses could help address this drug development challenge.
Discussion
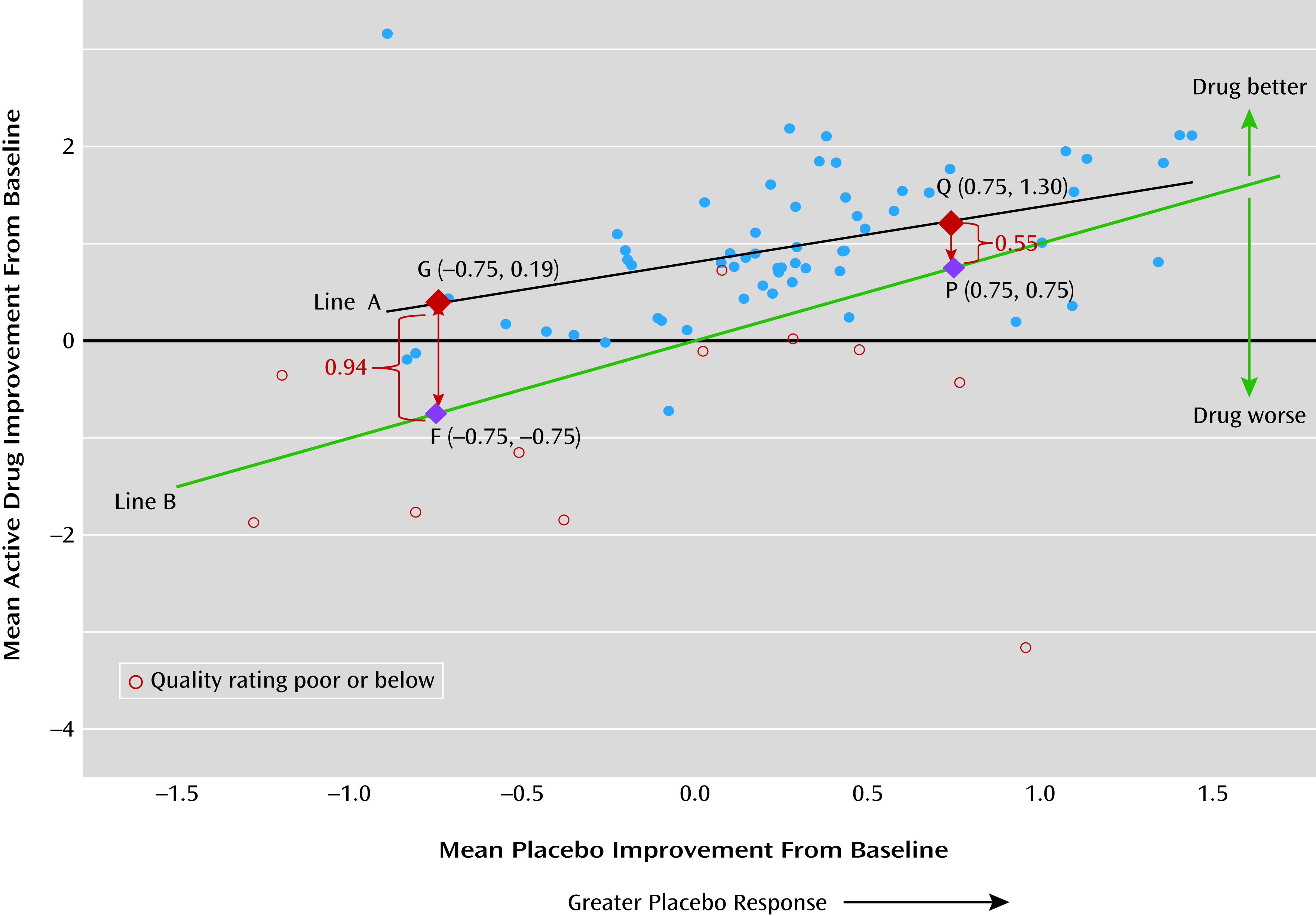
Our findings showed significant mean placebo improvement from baseline to endpoint (2 to 12 weeks), with a mean SMC of −0.33 (95% CI=−0.44, −0.22; N=50 studies). Smaller drug-placebo difference was associated with greater level of placebo response, and drug and placebo responses were positively correlated. Given the modest efficacy and tolerability issues associated with antipsychotics, a placebo improvement effect size of about 0.3 would make detecting drug efficacy signal challenging.
We also found a smaller drug-placebo difference in studies with greater placebo response (
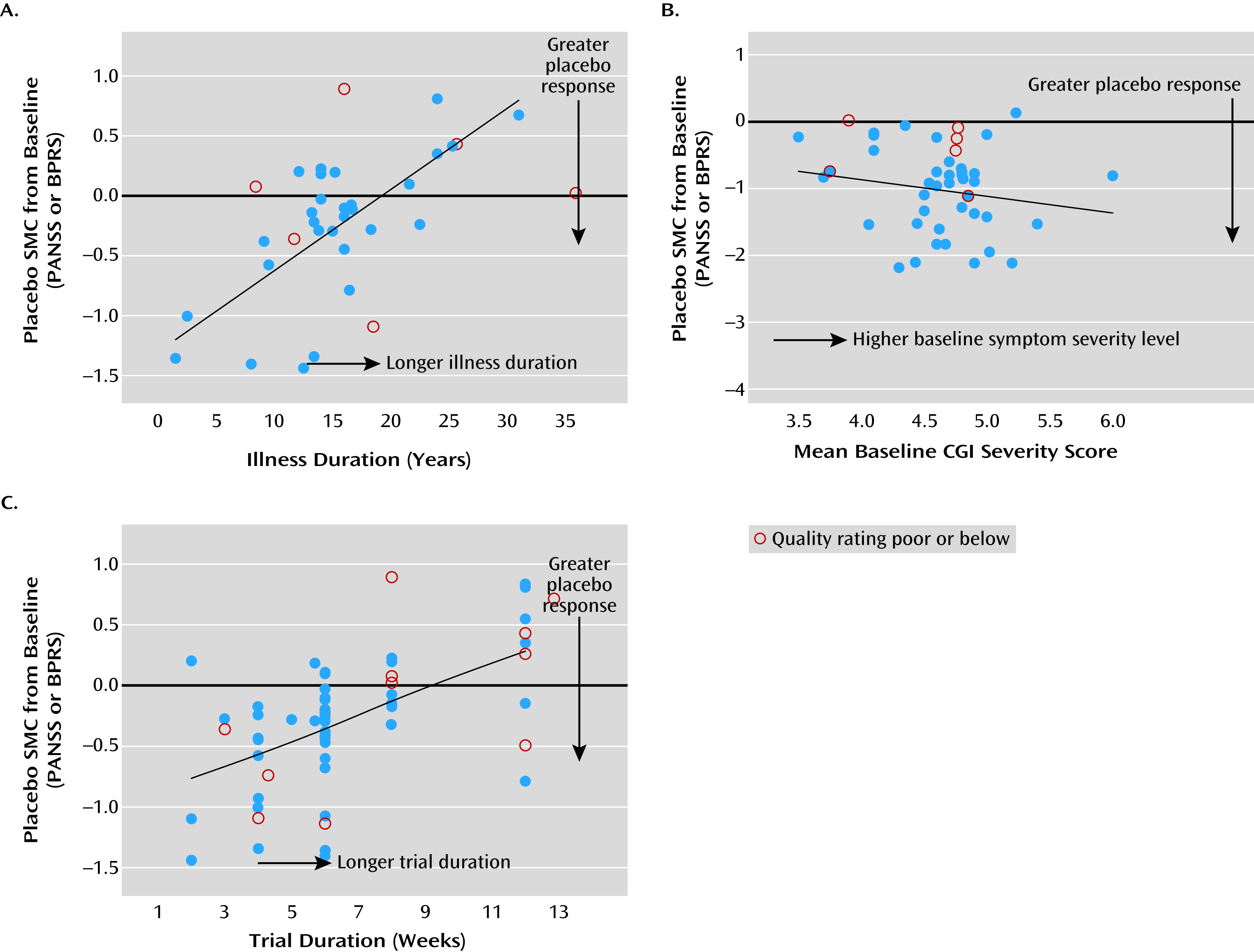
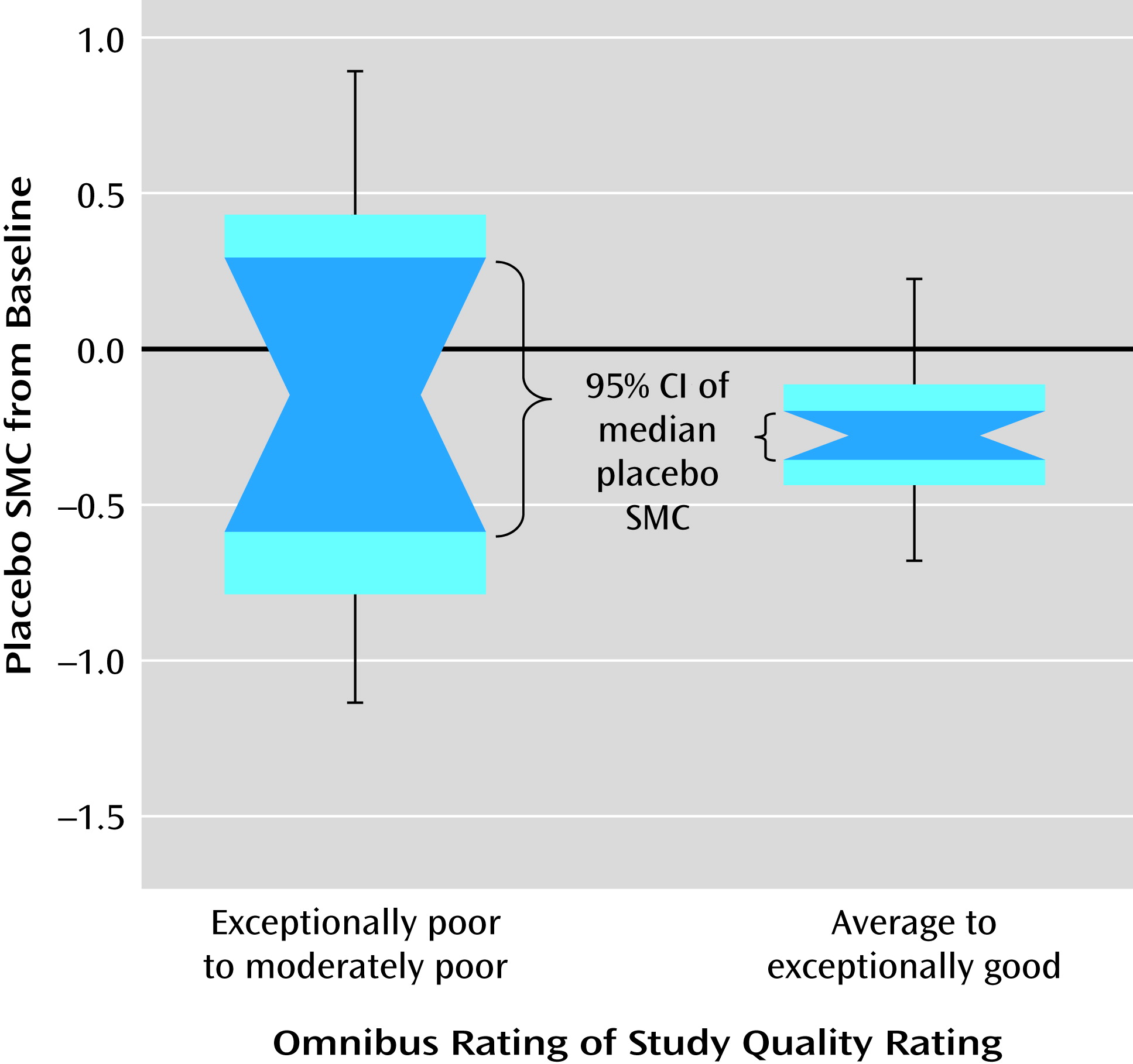
Figure 2). There were significant heterogeneities in the magnitude of placebo response and study quality across studies. Both placebo response and study quality increased significantly over time. A number of significant factors not previously empirically documented that affect placebo improvement effect size were identified. Lower age, shorter duration of illness, greater baseline symptom severity, and shorter trial duration were significantly associated with greater placebo response, while country (United States compared with others) did not, with or without statistical adjustment for study year influence. More study sites, fewer university or VA treatment settings, a lower percentage of participants assigned to receive placebo, or more study arms (if limited to studies since 1998) were associated with greater placebo response, but these were not independent of publication year. Study quality affected the variability but not mean levels of placebo response, thus affecting the reproducibility of effect size. To our knowledge, this is the first study to systematically evaluate the impact of study quality on the mean level as well as the variability of placebo response across trials.
We corroborated the previous observations that studies of shorter trial duration, lower age, or shorter duration of illness are vulnerable to substantial placebo response (
5,
7,
13). We also confirmed earlier findings that placebo response increased over the study years from 1993 to 2010 (
4,
5,
7,
21), and a greater placebo improvement was observed in studies with more study sites (
14,
16), fewer academic treatment settings (
18), and more study arms in studies since 1998 (
14). Greater placebo response was associated with higher mean baseline symptom scores (
5,
13), possibly as a result of a regression-to-mean effect (
32,
33). Interestingly, Chen et al. (
5) reported a significant association between change in score on the PANSS from baseline in both placebo and treated groups, with correlations at least two to three times higher in the treated groups. Improvement in study quality over time might reflect better trial methodologies or better reporting of randomized controlled trials, as evidenced by a reduction in the variability of the SMC from baseline. This could potentially improve the reproducibility of effect sizes across independent studies.
Our findings highlight the complex nature of the current trend of a rising placebo response, its association with various intrinsic factors, and correlated changes over time. The influence of study publication year and related period effects (before or after 1993–1997) on placebo response and study quality were significant, possibly reflecting a shift in patient population, trial methodology, trial execution, or reporting of psychiatric trials over the years. Because these constituted important sources of potential confounding effects, the identification of contributors to placebo response should be adjusted for study year and related period effects for less biased and more generalizable results across time. As shown in Figure S3 in the online
data supplement, observed placebo response patterns may be dependent on the time period and study year. Leucht et al. (
7), for example, included study publication year as a moderator in a meta-regression analysis of atypical antipsychotics compared with placebo and reported diminished drug-placebo differences in the reduction of symptom scores.
Because potential contributors to placebo response are generally not independent of one another, the choice of explanatory variables as determined by the usual stepwise method is generally unstable and can vary across samples and subsamples (
34). Furthermore, the stepwise method runs the inflated risk of detecting chance features in the data and can frequently lead to the selection of different predictors when applied to new data samples. This could especially be the case when the ratio of number of studies to number of predictors is small (
35), as is common in meta-regression analyses of aggregate data. In this study, we applied a covariate-adjusted regression approach to determine effect for individual predictor on placebo response, which remained stable even with different selections of correlated confounders (equation 2).
As shown in our analysis, the effect of trial duration was found to be consistent with four different sets of confounders. The regression coefficient for trial duration was 0.081 after accounting for study year as the only confounder in a parsimonious model; 0.079 after replacing study year with treatment setting and number of study sites as confounders; and 0.133 in the full model after accounting for study year plus all six other potential confounders. Notably, the estimated effect of trial duration on placebo response remained the same after dropping study year from the full model.
While study year can be considered a common marker associated with many important factors affecting placebo response, it is important to identify additional predictors above and beyond those influenced by study year. This meta-regression analysis found that age, duration of illness, baseline symptom severity, and trial duration had significant effects on placebo response, independent of the effect of study year.
Various intrinsic factors may influence the trend of rising placebo response. For example, the more noticeable extrapyramidal symptoms associated with conventional antipsychotics conceivably could make it easier for investigators or study participants to guess treatment assignment, thus attenuating placebo response. Patients with a shorter duration of illness may be more susceptible to placebo response because of heightened expectations based on treatment earlier in the course of illness. Greater placebo response among those with higher baseline positive or total symptom severity may reflect the nonspecific benefits of receiving more frequent assessments and attention in a trial (
36) or the immediate stabilization effect of hospitalization, as required in many studies (
37).
Limitations of our analysis include the use of response data from the placebo group alone in the meta-regression analysis. Although drug-placebo difference constitutes the direct measure of efficacy signals, placebo response has been shown to be the major cause underlying the diminished differences between drug and placebo responses (
21). This in turn contributes to the rising failures of randomized controlled trials in schizophrenia (
4–
10,
21). Our findings showed the magnitude of placebo response to be positively associated with the magnitude of drug response, but the magnitude of drug-placebo differences were not the same for all placebo response levels. While there have been many thorough reviews of mean drug-placebo difference, only limited data are available on the evaluation of placebo response itself, and with inconclusive results. There is a tremendous need for a thorough review of placebo response without confounding by active drug effects. Because the mechanisms of action, tolerability, and side effect profiles differ markedly among antipsychotic agents, drug response is a major determinant of the magnitude of drug-placebo differences. This analysis incorporated studies involving 23 different antipsychotic agents. Investigations of their respective contributions to the efficacy signals are useful but need to be addressed by a patient-level meta-analysis (
38). Finally, the use of a pooled active drug arm (from 146 active treatment arms on 23 antipsychotic agents in this database) as one control group would add substantial noise to the analysis of drug-placebo response.
In summary, this meta-analysis identified a broader range of study design parameters as potential contributors to placebo response in clinical trials of antipsychotic drugs in schizophrenia, and perhaps in psychopharmacology trials in general. Our findings argue for trials to be longer in duration, with a minimum period of 6 weeks, as well as for caution when expanding the number of investigative sites beyond 40, especially in nonacademic settings with limited clinical research experience or when incorporating unusually high baseline severity in study inclusion criteria (CGI severity score >5) (
33). Our results also suggest that greater consideration be given to study design factors, such as the number of treatment arms, features of investigative sites, and treatment settings, as well as to patient characteristics, such as illness duration.