Although cognitive-behavioral therapy (CBT) is a first-line treatment for panic disorder with agoraphobia (
1), little is known about the neural substrates underlying treatment response. Fear conditioning represents a central pathway for the development and maintenance of panic disorder with agoraphobia (
2–
4). Behavioral studies have found altered safety signal processing (
5,
6) and enhanced resistance to extinction (
7) in patients with panic disorder. In line with this, neuroimaging studies have indicated altered activation in the right anterior cingulate cortex, the amygdala, and the brainstem in patients with panic disorder during instructed fear conditioning (
8), with patients exhibiting increased activation during a safety condition. These findings indicate that a network that signals fear is incorrectly activated in individuals with panic disorder with agoraphobia.
Neuroimaging studies of treatment-related neural changes in patients with panic disorder are rare but provide the first evidence that brain activation is sensitive to change, although results remain highly inconsistent (
9–
12). It also remains unknown how changes in basic brain metabolism or brain metabolism at rest relate to dysfunctional processes of interest (e.g., fear conditioning). A recent analysis of treatment effects (
3), based on the sample in the present study, found neural correlates of enhanced differential conditioning in the left inferior frontal gyrus that attenuated after CBT. Despite this significant reduction, there was increased functional connectivity between the inferior frontal gyrus and limbic structures (e.g., the amygdala and the hippocampus) that remained stable across time. Although these findings contribute to our understanding of the pathophysiology of panic disorder, neural differences among patients determining the effectiveness of CBT and pathways of neuroplastic change underlying successful treatment response have not yet been evaluated.
Given previous evidence for the relevance of altered fear conditioning, safety signal processing, and fronto-limbic connectivity in patients with panic disorder with agoraphobia, we investigated 1) the predictive value brain activation patterns during fear conditioning have for treatment response and 2) changes in brain activation associated with treatment response. We hypothesized that responders and nonresponders would differ 1) in neurofunctional activation in neural networks subserving fear processing (e.g., the amygdala, hippocampus, and anterior cingulate cortex) during safety signal processing, 2) in functional connectivity between fronto-limbic networks, and 3) in pathways of neuroplastic change after CBT.
Discussion
Despite increasing evidence on the neurobiology of panic disorder (
25,
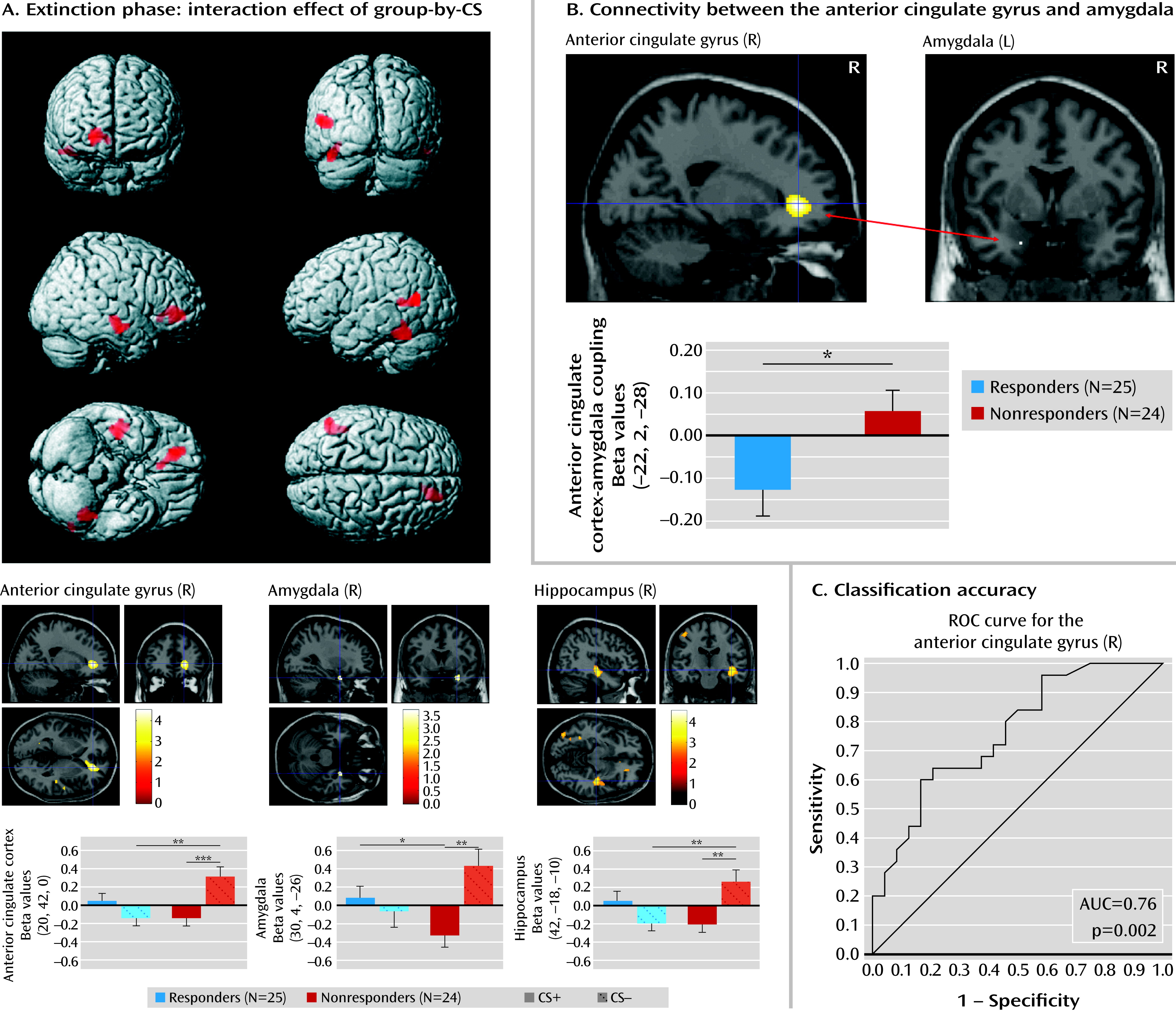
26), there is a lack of knowledge about the predictive value of neural activation patterns for therapeutic outcome. We focused on neural correlates of fear conditioning and neuroplastic changes after CBT as a marker of the pathophysiology of panic disorder with agoraphobia and putative pathways of change. Patients who did not respond to treatment exhibited enhanced activation in the pregenual anterior cingulate cortex, the amygdala, and the hippocampus during safety signal processing compared with responders. These increased activations among nonresponders normalized after CBT, while treatment response was associated with an increase in hippocampal activation when processing stimulus contingencies. Nonresponse was further associated with a lack of inhibitory functional anterior cingulate cortex-amygdala coupling that did not change after CBT.
Fear conditioning has been utilized as an experimental approach to better understand pathological forms of anxiety. It has been demonstrated to correlate with activity in a neural network encompassing the amygdala, the hippocampus, and the anterior cingulate cortex in the human brain (
27). Our study is one of the first, to our knowledge, to apply fear conditioning as a neurofunctional marker of treatment response in patients with panic disorder with agoraphobia. At baseline, enhanced activation in the above-described network in nonresponders was observed during the processing of stimuli that signal safety. These findings not only corroborate results of behavioral (
5,
6) and neuroimaging (
8) studies of altered safety signal processing in persons with panic disorder, but also show that activation of a neural network signaling fear in response to harmless stimuli is associated with treatment nonresponse. This may imply that patients with a detection bias toward threat, not differentiating safe versus unsafe contexts, do not sufficiently benefit from exposure-based treatment. Receiver operating characteristic curve analyses yielded good classification accuracy of neural activation patterns for treatment response, although the predictive value must be tested in a second, independent sample.
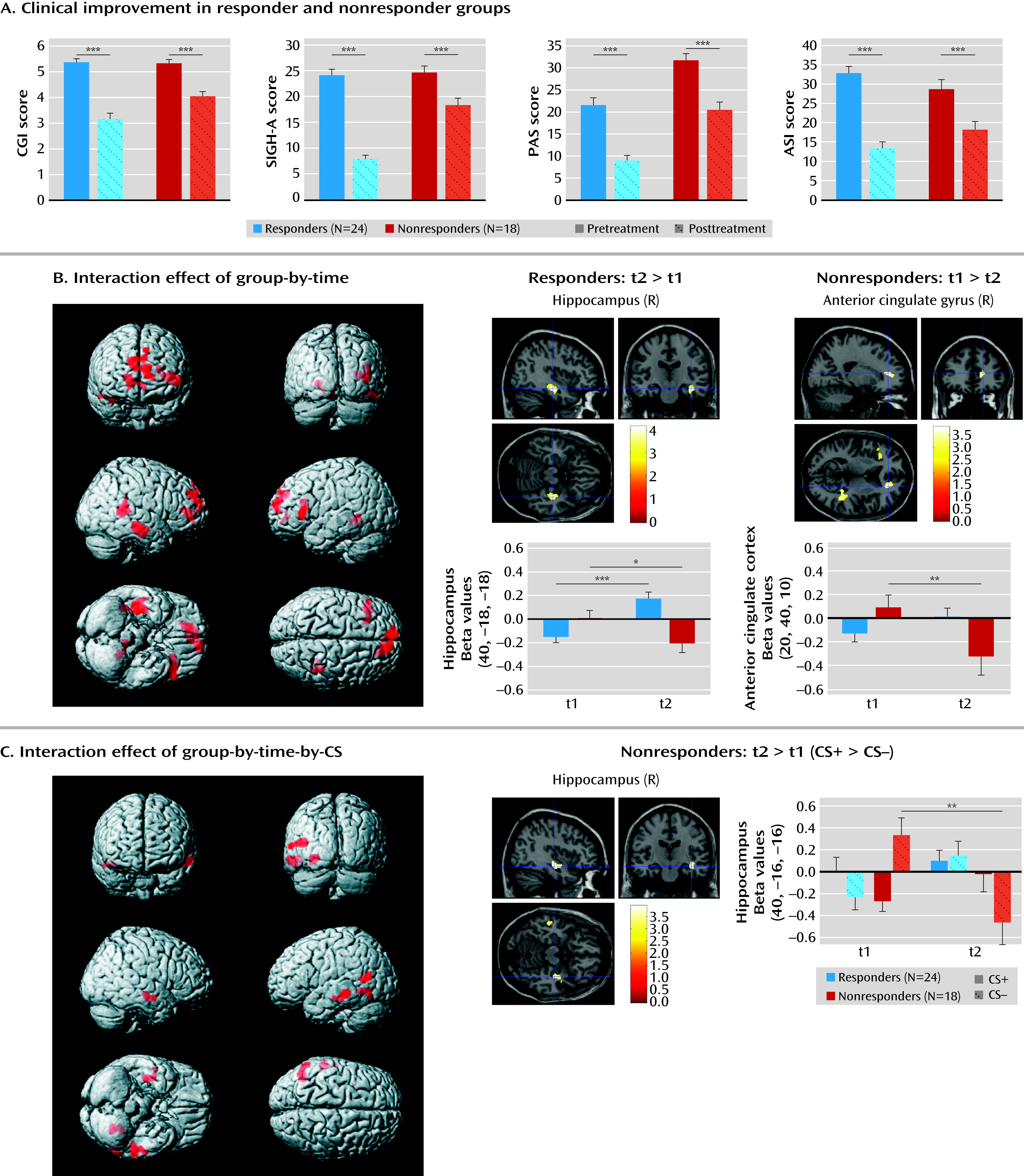
Our findings also indicated, however, that the dysfunctional baseline activations in patients who did not respond to treatment resolved after CBT, as indicated by reduced activity in the right hippocampus and anterior cingulate cortex. Although these patients were classified as nonresponders, they exhibited significant symptom reduction, indicating that they improved after CBT but not as much as patients who were classified as responders. Dysfunctional predispositions, as reflected by a neural bias toward threat detection, might disadvantage certain patients in therapy. In contrast, treatment response was associated with enhanced hippocampal activation when processing stimulus contingencies of both the CS+ and CS–. Brain lesion studies have emphasized the role of the hippocampus in conscious contingency awareness (
28) during fear conditioning. Since exposure therapy has been reported to involve strong conscious components (
29), we assume not only that sustained behavioral and neural changes after CBT require the unlearning of emotional responses, but also that this is most effective when contingencies between contexts, stimuli, and individual experiences are consciously learned and reappraised, as may be reflected by hippocampal involvement.
In a previous fMRI study (
9), changes of brain activation patterns in nine patients with panic disorder following short-term inpatient psychodynamic treatment were investigated using an emotional linguistic go/no-go task. Increased activation in the hippocampus and amygdala and low activation in the prefrontal cortex normalized after symptom improvement, but it is unclear whether these activation patterns can be generalized to the process of fear conditioning. Previous analyses of a subset of the present sample investigated the overall effect of CBT using a fear-conditioning task, comparing patients with healthy subjects. Enhanced activation in the left inferior frontal gyrus attenuated over time after CBT (
3). In line with a function of the inferior frontal gyrus in cognitive appraisal of negative emotions and threat (
30), results were interpreted as a reduction of negative cognitions following treatment. Supplementing these global effects of CBT, our data suggest that the amount of treatment success is modulated by additional neural circuits, such as medial prefrontal-limbic networks. Although aberrant activation in the amygdala could be identified as a baseline characteristic of nonresponse, we did not observe treatment-related changes in this structure. Findings evidencing general hyperactivation in the amygdala in persons with panic disorder have been inconsistent (
31) and may apply more to state than trait characteristics (
25).
Anterior cingulate cortex/medial prefrontal cortex-amygdala interactions have been implicated in fear extinction, emotion regulation, and trait anxiety (
32,
33). Successful fear extinction has been ascribed to inhibitory top-down modulation of the amygdala through medial prefrontal cortex inputs (
34), and functional connectivity between the amygdala and ventromedial prefrontal cortex has been observed during fear extinction and emotion regulation in humans (
35). Pezawas et al. (
32) reported a functional distinction between pregenual and subgenual components of the anterior cingulate cortex, with the former being negatively coupled with the amygdala, while the latter exhibited a positive coupling. The anterior cingulate cortex seed region we used in this study is located in the pregenual area, thus corroborating the notion of an inhibitory relationship to the amygdala. As shown in our results, this brain circuit is functionally relevant for treatment response in exposure-based CBT: responders were already characterized by a relatively higher inhibitory connectivity in this circuit before treatment. The chance to benefit from exposure in which extinction learning is conveyed through medial prefrontal cortex/anterior cingulate cortex-amygdala interactions may be increased in those patients who already have a relatively strong inhibitory coupling between these structures before therapy. Replicating previous findings of stable fronto-limbic connectivity in this sample (
3), we did not observe significant changes over time. This may indicate that the observed pattern of connectivity represents either a vulnerability to or a trait factor for panic disorder with agoraphobia, a hypothesis that could be tested in high-risk samples. Alternatively, changes in functional connectivity may require therapeutic interventions of a longer duration than we used in this study.
There are several limitations to this study. About one-third of patients initially scanned could not be considered for the analysis. The subsample examined was, however, clinically comparable to both patients who dropped out of the fMRI study and those in the non-fMRI sample. Comorbid diagnoses were not excluded per se, since presence of comorbid depression or anxiety conforms to what is usually seen in practice and thus may improve the external validity of the sample. The number of diagnoses, particularly depressive disorders, was comparable between responders and nonresponders. We included depression scores as a covariate in the model to account for psychopathology that was not specific to panic disorder with agoraphobia. Autonomic indices of fear conditioning were not available for our sample because of site-specific technical restrictions, but a pilot study indicated successful fear conditioning during the task (
21). No further markers are available to support the hypothesis of altered safety signal processing. Finally, the study design is lacking an extinction recall phase. Main effects were observed in the extinction learning phase but not in the acquisition phase, making it difficult to distinguish between processes related to the recall of the conditioned response and the gradual induction of extinction. Including a familiarization phase that preceded the acquisition phase most likely induced latent inhibition (an effect in which preconditioning exposure to the CS delays subsequent conditioning), thus possibly shifting the recall of conditioned responses further into the extinction phase. Future studies should more closely address extinction deficits in this patient population (
7) using tasks that allow for a separate analysis of extinction learning and recall.
In summary, this study identified a brain network associated with treatment response in patients with panic disorder with agoraphobia. Altered safety signal processing and impaired inhibitory anterior cingulate cortex-amygdala coupling that will augment, rather than down-regulate, fear-circuit reactivity may represent an important baseline characteristic that predisposes a subgroup of patients to obtain less benefit from CBT. Our findings can guide future add-on approaches, such as repetitive transcranial magnetic simulation (
36) or neurofeedback (
37), to purposefully influence “disadvantageous” brain activity in patients who do not sufficiently respond to CBT. While this dysfunctional baseline pattern partly resolved after CBT, treatment response was characterized by neuroplastic change in the hippocampus, possibly indicating conscious encoding strategies. These findings may not only contribute to a better understanding of how neurofunctional predispositions interact with behavioral treatments in these patients, but they may also enlarge our knowledge about the pathways by which successful CBT is conveyed.
Acknowledgments
Principal investigators with respective areas of responsibility in the MAC study are as follows: V. Arolt (overall MAC program coordination; Münster, Germany); H.U. Wittchen (principal investigator for the randomized clinical trial and manual development; Dresden, Germany); A. Hamm (principal investigator for psychophysiology; Greifswald, Germany); A.L. Gerlach (principal investigator for psychophysiology and panic subtypes; Münster, Germany); A. Ströhle (principal investigator for experimental pharmacology, Berlin); T. Kircher (principal investigator for functional neuroimaging; Marburg, Germany); and J. Deckert (principal investigator for genetics; Würzburg, Germany). Additional site directors in the randomized controlled trial component of the program are as follows: G.W. Alpers (Würzburg, Germany); T. Fydrich and L. Fehm (Berlin-Adlershof, Germany); and T. Lang (Bremen, Germany).
The study sites and staff members are as follows: Greifswald, Germany (coordinating site for psychophysiology): Christiane Melzig, Jan Richter, Susan Richter, and Matthias von Rad; Berlin-Charité, Germany (coordinating center for experimental pharmacology): Harald Bruhn, Anja Siegmund, Meline Stoy, and André Wittmann; Berlin-Adlershof, Germany: Irene Schulz; Münster, Germany (overall MAC program coordination, genetics, and functional neuroimaging): Andreas Behnken, Katharina Domschke, Adrianna Ewert, Carsten Konrad, Bettina Pfleiderer, Christina Uhlmann, and Peter Zwanzger; Münster, Germany (coordinating site for psychophysiology and subtyping): Judith Eidecker, Swantje Koller, Fred Rist, and Anna Vossbeck-Elsebusch; Marburg and Aachen, Germany (coordinating center for functional neuroimaging): Barbara Drüke, Sonja Eskens, Thomas Forkmann, Siegfried Gauggel, Susan Gruber, Andreas Jansen, Thilo Kellermann, Isabelle Reinhardt, and Nina Vercamer-Fabri; Dresden, Germany (coordinating site for data collection, analysis, and the randomized controlled trial): Franziska Einsle, Christine Froehlich, Andrew T. Gloster, Christina Hauke, Simone Heinze, Michael Hoefler, Ulrike Lueken, Peter Neudeck, Stephanie Preiss, and Dorte Westphal; University of Würzburg, Department of Psychiatry (coordinating center for genetics): Andreas Reif and Caro Gagel; University of Würzburg, Department of Psychology: Julia Duerner, Hedwig Eisenbarth, Antje B. M. Gerdes, Harald Krebs, Paul Pauli, Silvia Schad, and Nina Steinhäuser; Bremen, Germany: Veronika Bamann, Sylvia Helbig-Lang, Anne Kordt, Pia Ley, Franz Petermann, and Eva-Maria Schroeder. Additional support was provided by staff from the coordinating center for clinical studies in Dresden, Germany: Xina Graehlert and Marko Käppler.
The randomized controlled trial was approved by the Ethics Committee of the Medical Faculty of Technische Universität, Dresden, Germany (project number, EK 164082006). The neuroimaging components were approved by the Ethics Committee of the Medical Faculty of Rheinisch-Westfaehlische Hochschule University, Aachen, Germany (project number, EK 073/07), and at all local sites. The experimental pharmacology study was approved by the Ethics Committee of the state of Berlin (EudraCT, 2006-00-4860-29).