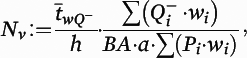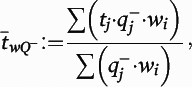In 1982, Feinberg hypothesized that increased pruning of existing synapses during adolescence may contribute to the onset of schizophrenia (
1), the incidence of which peaks toward the end of this developmental stage. Since then, increased synaptic pruning has become a major hypothesized pathogenic mechanism in schizophrenia, supported by the linking of two well-established observations. First, progressive gray matter loss has been observed in patients around the time of disease onset (
2). Second, significant reductions in the density of dendritic spines on pyramidal neurons, the major postsynaptic sites of excitatory, glutamatergic synapses in the cerebral cortex, have consistently been observed in schizophrenia subjects in multiple brain regions within the frontal and temporal neocortex (
3). As dendritic structural features are critical for signal processing (
4–
7), spine loss may then directly contribute to schizophrenia symptoms (
3,
8).
Dendritic spines on cortical pyramidal neurons are highly plastic throughout all stages of development, with shifting rate constants of formation and loss governing the net spine population in each developmental epoch. Spine and synapse formation is most robust in the perinatal period, leading to a peak in synapse numbers at 2–4 years of age. These excess synapses are then “pruned,” most intensely during adolescence, to adult levels. Throughout adulthood, large fractions of synapses are stably maintained, the physical incarnation of accumulated skills and memories (
9–
12). There is some turnover as skills and memories are lost, gained, or modified (
13–
15). In vivo imaging studies have confirmed the long-held view that adolescent pruning of dendritic spines is largely driven by elevated rates of elimination of mature or stable spines, while spine formation rates are similar to those seen in adulthood (
14).
In vivo imaging studies have also found that mature or stable spines are larger, while new or transient spines are smaller (
13,
15). Thus, the increase in synaptic pruning proposed to drive spine loss in schizophrenia should result in a deficit of larger spines in the adult cortex. Alternatively, recent genetic studies have implicated
N-methyl-
d-aspartate, brain-derived neurotrophic factor, calcium channel signaling, and mechanistic target of rapamycin signaling, which are pathways that are involved in spine formation and stabilization (
16,
17). If spine loss in schizophrenia is driven by a decrease in the rate of new spine formation and stabilization, a decrease in the number of smaller spines would be expected. To test these alternate hypotheses, we used the colocalization of the postsynaptic density (PSD) protein spinophilin and the cytoskeleton protein filamentous-actin (F-actin) to assess, for the first time, both the density and volume of dendritic spines in schizophrenia.
Method
Human Subjects
We examined tissue from two cohorts (
Table 1; see also Supplemental Table 1 in the
data supplement that accompanies the online edition of this article), the first comprising 20 subjects diagnosed with schizophrenia or schizoaffective disorder (together referred to as schizophrenia), and the second comprising 20 comparison subjects matched on the basis of sex and as closely as possible for age, postmortem interval, and handedness (
18–
21). We have previously reported on spine density, but not spine density by spine volume, in these cohorts (
22).
Human Tissue Processing
At the University of Pittsburgh brain bank, the right and left hemispheres of postmortem brain are processed differently. Tissue blocks from the left hemisphere are fixed in paraformaldehyde, while blocks from the right hemisphere are fast-frozen in isopentane. Thus, tissue from the left hemisphere was used for immunohistochemistry, while tissue from the right hemisphere was used for targeted mass spectrometry.
Immunohistochemistry
Tissue from the 20 pairs was divided into an initial cohort and a replication cohort: cohort 1 (N=12 pairs) and cohort 2 (N=8 pairs) (see Supplemental Table 2 in the online data supplement). These two cohorts were assayed independently. Tissue from each cohort was processed together across a series of immunohistochemical runs. To visualize dendritic spines, we used two markers in combination: a polyclonal antibody directed against spinophilin (Millipore AB5669, Billerica, Mass.), a protein that is highly enriched in spine heads, and the F-actin binding mushroom toxin phalloidin (Invitrogen A12380, Carlsbad, Calif.), which is also highly enriched in dendritic spines.
Image Collection and Processing
Matched pairs from each cohort were imaged during the same session by an experimenter who was blind to diagnostic group. All images were taken using a confocal microscope equipped with a 60× oil supercorrected objective (equipment details are available in the online data supplement). Tissue thickness was measured at each sampling site and did not differ by diagnostic group (F=0.05, df=1, 18.5, p=0.83) or cohort (F=2.35, df=1, 18.6, p=0.14). Image stacks were taken beginning 12.5 µm below the tissue surface closest to the cover glass, stepping up 0.25 µm with each image until the tissue surface was reached. This procedure produced an image stack of 50 individual planes, each 512×512 pixels in size. Exposure times for 488-nm and 568-nm excitation wavelengths were set to optimize the spread of the intensity histogram for each cohort and then were kept consistent for all subjects within the cohorts.
Images were processed using SlideBook, version 5.027, with keystrokes automated by a software program (Automation Anywhere, San Jose, Calif.). Camera background was subtracted from channels 488 and 568 prior to processing. Underlying gray level values were extracted from the mask objects. See the data supplement for more detail on image processing.
Calculation of Spine Density, Number, and Area
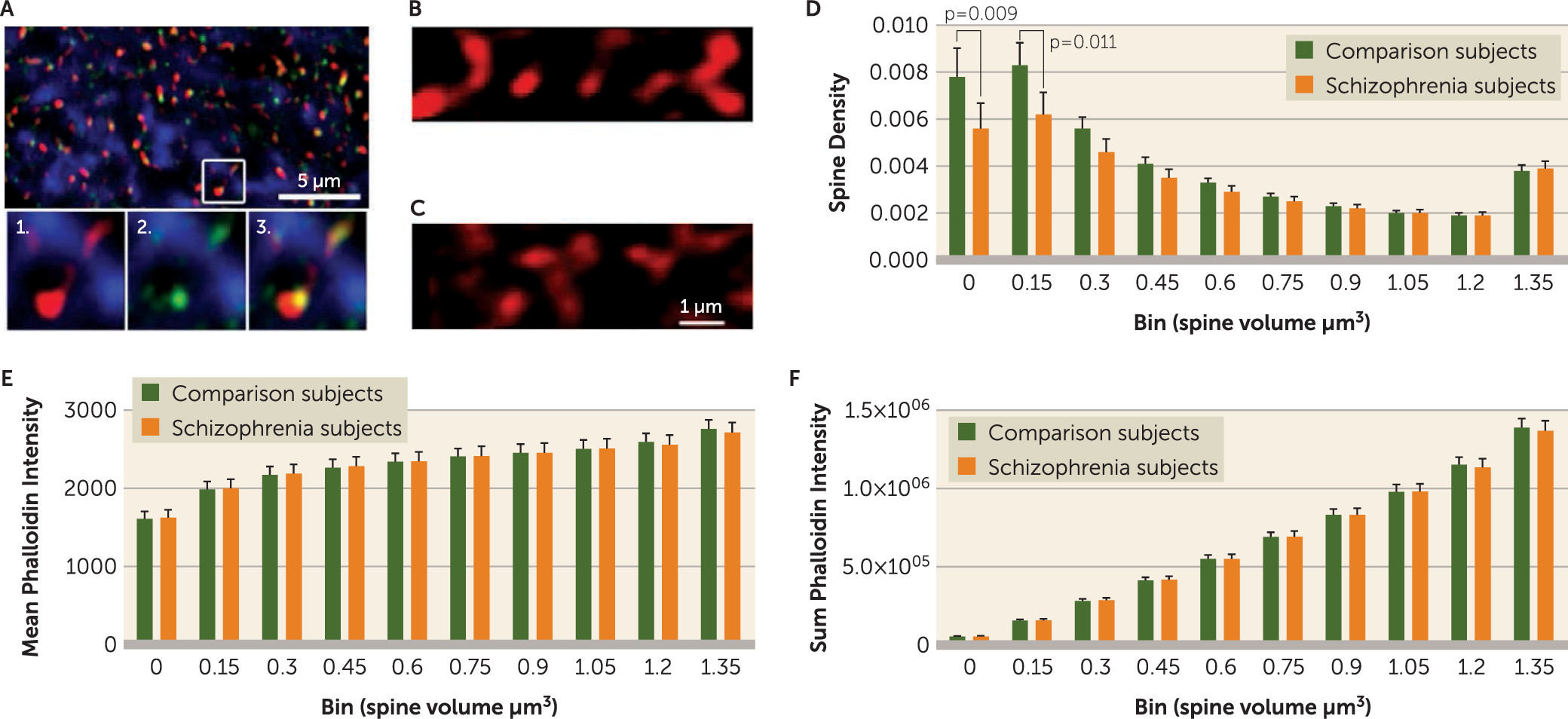
Although both spinophilin-immunoreactive and phalloidin binding are strongly localized to spines, each has some off-target label. Therefore, identification of putative dendritic spines required colocalization of spinophilin-immunoreactive and phalloidin label (
Figure 1), operationalized as phalloidin mask objects that overlapped (≥1 voxel) with a spinophilin-immunoreactive mask object. Spine density (N
v) in cohort 1 was calculated as previously described with minor modifications:
where
a is the area of the counting frames,
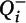
is the count of dendritic spines within the
ith block,
Pi is the count of the associated points hitting the region of interest in the
ith block,
h is the disector height (see the
online data supplement for additional details), and
BA is the cryostat block advance (50 μm for cohort 1 and 60 μm for cohort 2). In addition,
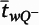
is the block-and-number-weighted mean section thickness calculated using this formula:
where
tj is the local section thickness measured centrally in the
jth sampling frame and
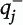
is the corresponding count of dendritic spines in the
jth frame. In addition,
wi is the block weight (i.e., either one or one-third). Because sections adjacent to the mapping sections were sampled for cohort 2, calculation of
Nv was made as described above but omitting the block weighting.
Targeted Mass Spectrometry
Tissue homogenates were prepared from fresh frozen human A1 gray matter, described above and in the
online data supplement. Total protein was extracted using SDS extraction buffer (0.125 M Tris–HCl [pH 7], 2% SDS and 10% glycerol) at 70°C. Protein concentration was measured using bicinchoninic acid assay (Micro BCA Protein Assay, Pierce). A pooled technical replicate sample composed of homogenate aliquots from all subjects was also prepared, and 20 μg of total protein from the gray matter homogenate or pooled sample was mixed with lysine
13C
6 stable isotope labeled neuronal proteome standard (
23) (
13C
6 STD; 20 μg) for on-gel trypsin digestion. Samples were organized in a block distribution. Each block was run on a single 10-well 4%−12% BisTris gel with two SeeBlue Plus2 prestained protein standards (Invitrogen). On-gel trypsin digestion was performed as previously described (
23), and samples were run 4 cm into the gel and divided into two fractions (above and below 65 K
d). Liquid chromatography–selected reaction monitoring mass spectrometry assay development and implementation was performed as previously described (
23,
24) (see the
data supplement for details).
Primary Neuronal Culture
Primary cortical neuronal cultures were prepared from embryonic day 17 Sprague-Dawley rats (Envigo and Charles River laboratories). Neurons were plated at 450,000 cells per well in 12-well plates. On day in vitro 12, the neurons were transfected with
CACNB4/Myc (OriGene; catalog number RR204310) and green fluorescent protein (GFP) (gift of Ryan Logan, University of Pittsburgh). Thus, in the same plate some neurons were transfected with both constructs and some with only one. Neurons were fixed on day in vitro 15 for imaging with mouse anti-c-Myc antibody (1:1000, monoclonal 9E10; Santa Cruz Biotechnology) or with mouse anti-
CACNB4 (1:100, monoclonal S10–7;
antibodies-online.com) and goat anti-c-Myc (1:100, polyclonal; Novus Biologicals). Image acquisition was performed on an Olympus (Center Valley, Pa.) BX51 WI upright microscope equipped with an Olympus spinning disk confocal (SDCM) using an Olympus PlanAPO N 10×0.40 NA air objective and a 1.42 numerical aperture 60× oil supercorrected objective.
Neurons were first categorized as either CACNB4-overexpressing or GFP-only controls based on c-Myc intensity. GFP-positive neurons on coverslips stained for both c-Myc and CACNB4 were imaged at 10×. Exposure times for the 488 channel were optimized, whereas the 568 and 647 channels were shot at fixed exposures of 447 ms and 3,000 ms, respectively.
Spine Counting and Masking
The TIFF files of all dendrites were imported into StereoInvestigator (MicroBrightField) for counting. Protrusions from the dendritic shaft were manually assigned to one of the following categories, blind to experimental condition: short mushroom spine, long mushroom spine, short stubby spine, long stubby spine, or filopodia. Criteria for assignment have been previously described (
25). Briefly, mushroom spines had distinct heads, while stubby spines did not. Long spines had a length greater than maximal width, and the rest were short. Filopodia were longer than 2 μm, thinner than 0.3 μm, and lacked a distinct head. For a given dendrite, the number of protrusions in each category was recorded, and the densities per μm of dendrite length for all spines, mushroom and stubby subtypes, and filopodia were calculated. A subset of the 60× dendritic images used for spine counting was randomly selected, blind to condition, for spine area analysis. In total, 35 GFP+ only dendrites and 38 Myc+/GFP+ dendrites were analyzed. Details on spine area calculation can be found in the
data supplement.
Statistical Analysis
Human spine density by volume.
The data were analyzed through a linear mixed model with the pair effect taken into account. Spine density was assumed to be normally distributed. In the model, pair, cohort, diagnosis, and spine size category were the fixed effects. Subject was treated as a normal random effect to account for the repeated measures within each subject. Insignificant interaction terms were not included in the final model. The Kenward-Roger method was used to adjust for the denominator degrees of freedom. The analysis was implemented in SAS, version 9.4, with the PROC MIXED procedure. Categorical confounding effects were also assessed (see the data supplement).
Human spine density continuous confounding effects.
For the analysis of continuous confounding effects, the percentage change of spine density within each pair was calculated as follows: ([C−S]/C) × 100%. A simple linear regression analysis was done on each of the two confounding variables separately (age at onset and duration of disease). The slope of the percentage change of spine density on each confounding variable was estimated, and the p value to test whether the slope was significantly different from zero was provided in the table.
Protein level correlations with spine density by size.
Pearson’s correlation was used to determine the relationship between peptide expressions and spine density by volume.
Discussion
We observed that spine decreases in deep layer 3 of the primary auditory cortex in schizophrenia were limited to spines of smaller volumes. The decrease in small spines most likely reflects a reduction in the number of new or transient spines. The appearance of new or transient spines and their probability of stabilization are regulated by local calcium signaling generated by voltage-gated calcium channels (
28,
29). Glutamate uncaging can also induce spinogenesis (
12,
30), and the presence of a PSD is essential, but not sufficient, for new spine stabilization at a bouton (
31). Genome-wide association (GWA) (
17) and rare variant (
16) analyses of schizophrenia have identified voltage-gated calcium channels and glutamate signaling from the PSD to the F-actin cytoskeleton in schizophrenia risk. It is important to note that the majority of small or new spines, even those that sample a bouton and acquire a PSD, are transient (
31). Thus, even in healthy circuits, the early stages of spine stabilization are tentative, and the genetic factors that converge upon and destabilize PSD and voltage-gated calcium channel activity would likely have their greatest effect in these early, less robust stages of the spine life cycle.
An important consideration regards the potential effects of postmortem interval. We note that the cohorts in the spine density analysis were matched for postmortem interval as closely as possible. Furthermore, we did not observe any correlation between postmortem interval and small spine density (see Supplemental Table 2 in the online data supplement), strongly suggesting that postmortem interval does not affect our findings in schizophrenia relative to comparison subjects. Nevertheless, it is possible that postmortem interval could alter the relationship between spine size and spine class (i.e., stable or mature compared with new or transient), affecting our interpretation of the nature of the reduction in small spines in schizophrenia.
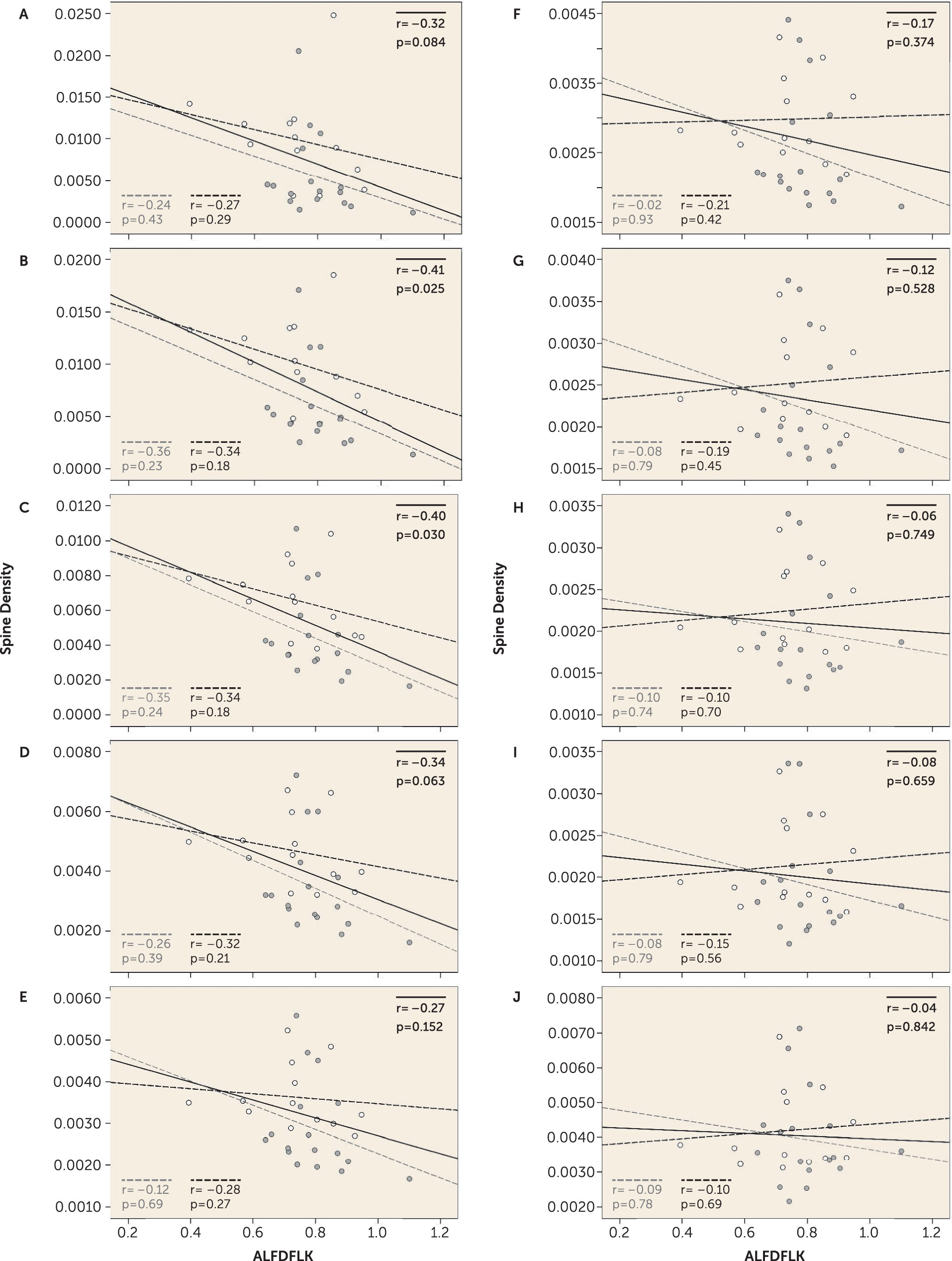
We sought to determine whether a link between PSD and/or voltage-gated calcium channel signaling proteins and small spine loss was present in these cohorts, finding that the expression of one tryptic peptide, ALFDFLK, was inversely correlated with the density of only smaller spines (
Figure 2). The amino acid sequence ALFDFLK is unique to the four calcium channel beta subunits (
CACNB1 through
CACNB4). Of these subunits,
CACNB2 and
CACNB4 have been implicated in schizophrenia pathogenesis by GWA (
17) and rare variant (
16) analyses. Of these two,
CACNB4 is the predominant isoform expressed in the adult temporal lobe (
32,
33). In line with this observation in human tissue, we found that the overexpression of
CACNB4 decreased the density of smaller spines in primary neuronal culture. However, as ALFDFLK is present in other
CACNB variants, it will be important to trace this effect to specific proteins.
CACNB4, like all voltage-gated calcium channel beta subunits, modulates the trafficking and biophysical properties of the alpha pore forming subunits (
34). While the association of common and rare alleles of several voltage-gated calcium channel subunits with schizophrenia risk is well established (
17,
35), a consensus on whether they result in a loss or gain of function, and on how they may exert an effect on synapses, is lacking (
35). Our findings suggest that
CACNB4 overexpression depresses the calcium currents that drive spine formation and/or stabilization. Previous studies provide some potential mechanisms and precedent for this observed effect.
CACNB proteins are required for voltage-dependent inactivation of calcium channels by a number of second messengers (
36). Moreover,
CACNB4 knockout mice display evidence of increased calcium currents and increased excitability, manifest as epilepsy, suggesting that the dominant effect of
CACNB4 expression may be to reduce calcium currents (
36).
CACNB4 is also noteworthy among the beta subunits in that it traffics from the synapse to the nucleus, affecting gene expression (
37) and providing a second mechanism by which voltage-gated calcium channel activity could affect spine plasticity in addition to the regulation of local calcium signaling.
Despite identifying a significant correlation between ALFDFLK and small spine density, ALFDFLK levels were not significantly increased in our schizophrenia subjects. However, our in vitro data firmly demonstrate that increased
CACNB4 expression alone can drive small spine loss. This combination of findings is consistent with the current model for schizophrenia risk in which multiple genetic variants and epigenetic and environmental influences are believed to act in concert. No single gene or protein is believed to account for more than a small share of the population risk. As
CACNB4 interacts with and regulates multiple voltage-gated calcium channel subunits, many of which are also implicated in schizophrenia (
17), it likely acts in concert with, or parallel to, other voltage-gated calcium channel genetic risk factors.
In the adult cortex, current evidence indicates that new or transient spines act as the substrate for reorganization of cortical networks in response to experience (
38). These spines accomplish this rewiring both by providing for the formation of new persistent synapses (
13,
15,
39) and by contributing to the destabilization of persistent synaptic spines through bouton competition (
31,
40). Thus, a decrease in the rate of spine formation and/or their subsequent stabilization would result not only in their depletion but also in a decreased turnover rate of large spines by decreasing competitive loss, potentially explaining the observed conservation of large spines in schizophrenia despite a decrease in the small spines from which they are formed. The predicted cognitive consequences of new or transient spine reductions, in a general sense, would be impairments known to affect individuals with schizophrenia: deficits in new learning (
41). Recently, auditory training exercises have shown promise for remediating auditory processing deficits in schizophrenia patients (
42). Impairments in the generation or stabilization of new or transient spines in some subjects with schizophrenia may limit the available gains through such an approach. Thus, identification of therapeutics promoting small spine generation and stabilization might enhance the benefits of such training, providing a measurable outcome of drug efficacy.
In summary, our finding that A1 layer 3 spine loss in schizophrenia is limited to smaller spines strongly suggests that adult spine deficits result from a failure to generate and/or stabilize new or transient spines. This finding requires a rethinking of the overpruning hypothesis and should spur a more in-depth investigation of the mechanisms that govern spinogenesis, spine stabilization, and their role in schizophrenia. As a first step, we have identified one such mechanism, CACNB4 expression levels, which could contribute in part to the loss of small spines in schizophrenia. Future studies to confirm the effect of CACNB4 and other CACNB subunits in vivo are warranted and might include direct observation of spine dynamics. In addition, the pleiotropic genetic associations of calcium channel subunits with schizophrenia risk highlight the continued need to expand investigations beyond single proteins to the larger signaling network to enhance understanding of how genetic risk is translated into synaptic pathology.