Identification of Developmental and Behavioral Markers Associated With Genetic Abnormalities in Autism Spectrum Disorder
Abstract
Objective:
Method:
Results:
Conclusions:
Method
Sample Collection
Genetic Data and Participants
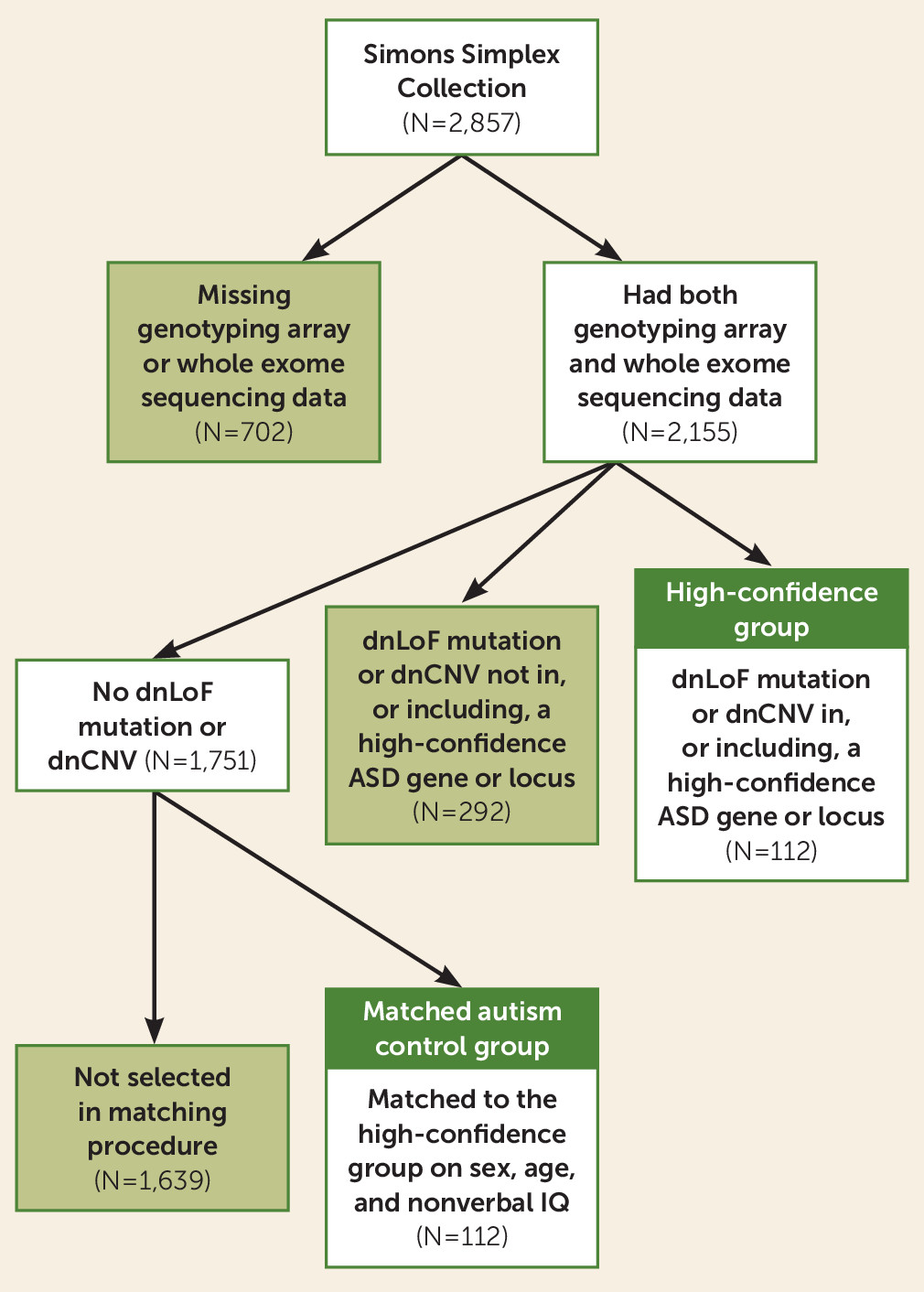
| Characteristic | None (N=1,751) | Low-Confidence Group (N=292) | High-Confidence Group (N=112) | Matched Autism Control Group (N=112) | ||||
|---|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | N | % | |
| Male | 1,546 | 88 | 245 | 84 | 86 | 77 | 86 | 77 |
| White | 1,375 | 79 | 224 | 77 | 95 | 85 | 86 | 77 |
| Hispanic | 211 | 12 | 36 | 9 | 9 | 8 | 14 | 13 |
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| Age (months) | 107.56 | 42.56 | 114.93 | 44.85 | 113.10 | 39.75 | 112.83 | 39.40 |
| Nonverbal IQ | 86.28 | 26.06 | 80.34 | 27.31 | 74.88 | 23.96 | 75.46 | 24.40 |
Measures
Statistical Analysis
Results
| Matched Autism Control Group Versus High-Confidence Groupb | Group-by-Nonverbal IQ Interaction (Nonverbal IQ as Moderator)b | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Measure | Pairs (N) | Matched Autism Control Group | High-Confidence Group | Test Statistic | p | FDR p | Test Statistic | p | FDR p | ||
| Mean | SD | Mean | SD | ||||||||
| Age (months) (matching variable) | 112 | 112.83 | 39.41 | 113.10 | 39.75 | ||||||
| Nonverbal IQ (matching variable) | 112 | 75.46 | 24.00 | 74.88 | 23.96 | ||||||
| Verbal IQ | 112 | 68.05 | 31.18 | 74.28 | 29.49 | –2.81 | 0.01 | 0.02 | 1.16 | 0.28 | 0.75 |
| Nonverbal IQ minus verbal IQ | 112 | 7.40 | 16.10 | 0.61 | 16.46 | 3.12 | 0.002 | 0.01 | 1.16 | 0.28 | 0.75 |
| Autism Diagnostic Interview–Revised | |||||||||||
| Age at first words (months) | 112 | 27.28 | 17.43 | 29.71 | 27.18 | –0.80 | 0.43 | 0.55 | 3.69 | 0.06 | 0.39 |
| Age at first phrases (months) | 112 | 49.10 | 23.16 | 46.86 | 31.81 | 0.71 | 0.48 | 0.58 | 2.56 | 0.11 | 0.50 |
| Age at onset of walking (months) | 112 | 13.54 | 3.44 | 15.79 | 4.93 | –4.28 | <0.001 | 0.001 | 12.13 | 0.001 | 0.02 |
| Vineland Adaptive Behavior Scales, 2nd Edition | |||||||||||
| Communication, standard score | 112 | 72.63 | 12.17 | 73.49 | 12.19 | –0.86 | 0.39 | 0.55 | 0.46 | 0.50 | 0.92 |
| Daily living skills, standard score | 112 | 74.25 | 12.77 | 72.81 | 13.22 | 1.11 | 0.27 | 0.43 | 0.06 | 0.81 | 0.97 |
| Socialization, standard score | 112 | 69.83 | 11.51 | 69.30 | 12.20 | 0.44 | 0.66 | 0.71 | 0.04 | 0.84 | 0.97 |
| Adaptive behavior composite, standard score | 112 | 70.82 | 10.53 | 69.72 | 10.94 | 1.15 | 0.25 | 0.42 | 0.01 | 0.93 | 0.97 |
| Peabody Picture Vocabulary Test, standard score | 109 | 74.86 | 31.24 | 81.77 | 25.23 | –3.32 | 0.001 | 0.01 | 10.93 | 0.001 | 0.02 |
| Purdue pegboard task | |||||||||||
| Both hands, raw score | 70 | 6.14 | 3.41 | 6.21 | 3.33 | –0.20 | 0.85 | 0.85 | 0.59 | 0.44 | 0.92 |
| Dominant hand, raw score | 70 | 8.81 | 2.78 | 9.04 | 3.10 | –0.64 | 0.52 | 0.61 | 0.10 | 0.75 | 0.96 |
| Nondominant, raw score | 70 | 7.77 | 3.50 | 8.39 | 3.59 | –1.38 | 0.17 | 0.34 | 0.11 | 0.74 | 0.96 |
| Child Behavior Checklist | |||||||||||
| Internalizing scale, total T score | 111 | 58.88 | 9.15 | 60.46 | 8.62 | –1.33 | 0.19 | 0.35 | 0.40 | 0.53 | 0.92 |
| Externalizing scale, total T score | 111 | 55.70 | 10.57 | 57.85 | 11.60 | –1.44 | 0.15 | 0.32 | 0.15 | 0.70 | 0.96 |
| Social Responsiveness Scale, total T score | 111 | 81.34 | 9.93 | 80.34 | 11.13 | 0.71 | 0.48 | 0.58 | 0.01 | 0.92 | 0.97 |
| Autism Diagnostic Interview–Revised | |||||||||||
| Social domain, total score | 112 | 22.43 | 5.35 | 20.15 | 5.43 | 3.43 | 0.001 | 0.01 | 0.18 | 0.67 | 0.96 |
| Nonverbal communication, total score | 112 | 10.01 | 3.47 | 9.09 | 3.34 | 2.25 | 0.03 | 0.07 | 1.11 | 0.29 | 0.75 |
| Restricted and repetitive behaviors, total score | 112 | 6.76 | 2.59 | 6.49 | 2.59 | 0.84 | 0.40 | 0.55 | 0.01 | 0.91 | 0.97 |
| Autism Diagnostic Observation Schedule | |||||||||||
| Total, calibrated severity score | 112 | 7.79 | 1.53 | 7.29 | 1.82 | 1.26 | 0.21 | 0.37 | 0.54 | 0.46 | 0.92 |
| Social affect, calibrated severity score | 109 | 7.47 | 1.69 | 7.07 | 1.84 | 2.23 | 0.03 | 0.07 | 2.48 | 0.12 | 0.50 |
| Restricted and repetitive behaviors, calibrated severity score | 109 | 8.18 | 1.76 | 7.86 | 1.98 | 1.65 | 0.10 | 0.25 | 2.38 | 0.12 | 0.50 |
| Overall ASD diagnostic certainty | 112 | 13.83 | 1.98 | 12.55 | 2.60 | 4.25 | <0.001 | 0.001 | 1.82 | 0.18 | 0.62 |
| N | % | N | % | ||||||||
| Autism Diagnostic Observation Schedule modulec | 112 | 8.63 | 0.003 | 0.01 | 0.20 | 0.65 | 0.96 | ||||
| 1 | 31 | 28 | 19 | 17 | |||||||
| 2 | 24 | 21 | 20 | 18 | |||||||
| 3 | 53 | 47 | 71 | 63 | |||||||
| 4 | 4 | 4 | 2 | 2 | |||||||
| High ASD diagnostic certaintyd | 112 | 97 | 87 | 71 | 63 | 13.20 | 0.003 | 0.003 | 0.18 | 0.67 | 0.96 |
| Family history of major psychiatric problemse | 96 | 45 | 47 | 43 | 45 | 0.10 | 0.76 | 0.79 | 0.94 | 0.33 | 0.77 |
| Seizures | 112 | 6 | 5 | 13 | 12 | 2.45 | 0.12 | 0.28 | 4.34 | 0.04 | 0.35 |
| Language deficit | 112 | 40 | 36 | 24 | 21 | 7.24 | 0.01 | 0.02 | 0.00 | 0.97 | 0.97 |
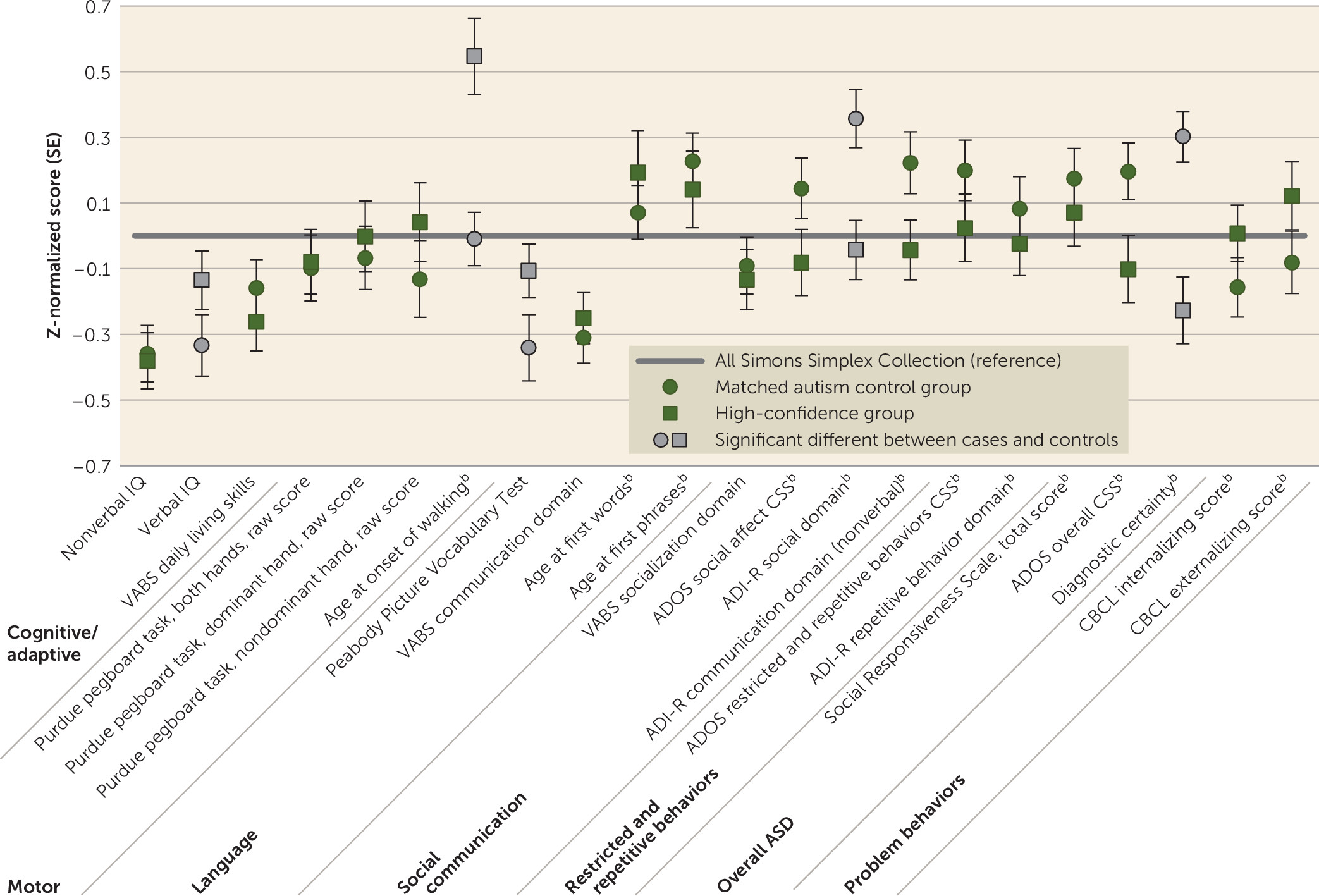
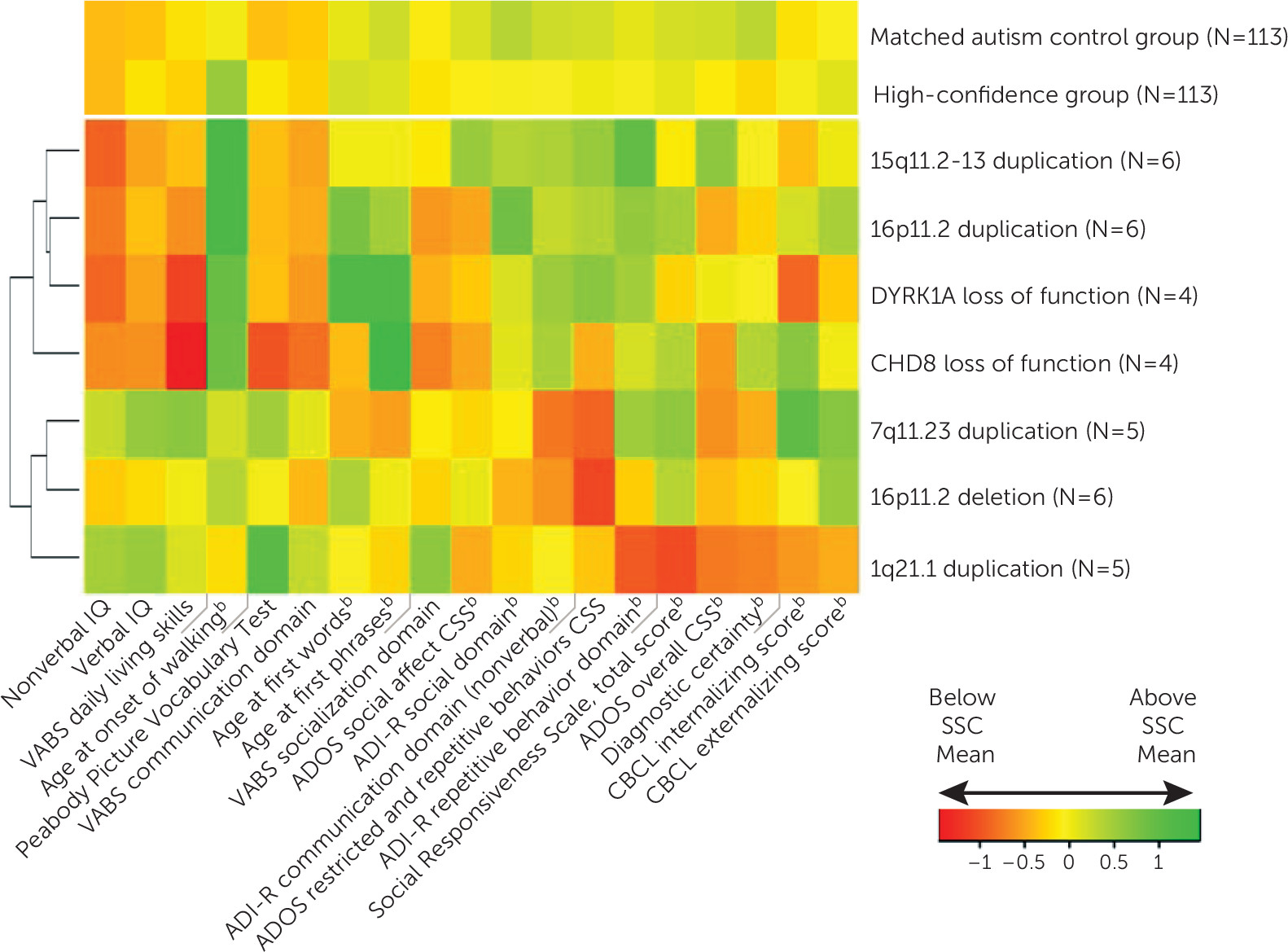
Discussion
Limitations and Future Directions
Supplementary Material
- Download
- 28.79 KB
- View/Download
- 355.16 KB
References
Information & Authors
Information
Published In


History
Keywords
Authors
Funding Information
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBLogin options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).