Genetic susceptibility to schizophrenia is highly polygenic, including many associated loci of small effect (
1,
2). Although individual risk alleles may convey an odds ratio of 1.1 or lower, the combination of all such effects across the genome holds substantial explanatory power. For example, any individual can be characterized by a polygenic risk score (PRS), representing the total number of risk alleles he or she carries, weighted by the odds ratio associated with each allele as derived from previous genome-wide association study (GWAS) findings (
3,
4). Although a high PRS for schizophrenia is not deterministic, PRSs derived from the Psychiatric Genomics Consortium (
1) account for approximately 7% of variation in the risk for schizophrenia (as measured on the liability scale [
5]), with about half of that variance accounted for by the top (genome-wide significant) loci. Additionally, individuals scoring in the top decile are approximately 15 times more likely to manifest the illness compared with those in the bottom decile (
1).
Given the explanatory power of PRS for susceptibility to schizophrenia, it is reasonable to ask whether these scores can be informative regarding clinical heterogeneity within the disorder (
2). For example, while antipsychotic drugs are the mainstay therapy for schizophrenia (
6,
7), up to 30%−40% of patients do not respond to antipsychotic treatment (
8), and many patients discontinue their medications due to lack of efficacy (
9). There is currently a paucity of clinically informative biomarkers, and pharmacogenomics is one approach to identifying predictors of treatment response (
10). To date, candidate-gene studies and a small number of GWAS have had limited success in identifying genetic variants replicably associated with antipsychotic treatment response. Thus far, only two variants (at the
DRD2 gene and the
COMT gene) have demonstrated consistent effects across multiple cohorts as demonstrated by meta-analysis (
11,
12). Although promising, their effect sizes are relatively small (odds ratios, 1.54 and 1.37, respectively), and predictive power is limited (
13).
Given previous findings suggesting that a family history of schizophrenia may be associated with poor clinical response (
14,
15), patients with higher genetic burden of schizophrenia may have poorer clinical outcomes. Compared with candidate gene approaches, PRS methods may better capture the full genomic underpinnings of illness and improve clinical prediction, as has been recently demonstrated with prostate cancer, in which higher PRS was associated with more aggressive illness (
16). One recent schizophrenia study utilized clinically assigned clozapine therapy as a proxy for treatment resistance (by comparing patients treated with clozapine with those who had never been prescribed clozapine) and found that the PRS was significantly higher among patients in the clozapine group compared with patients in the nonclozapine group (
17), although another study failed to replicate the finding (
18). However, both were cross-sectional studies that can be affected by ascertainment bias and inaccuracies of classification. For example, a similar cross-sectional study providing evidence for a pharmacogenetic role for the
BDNF Val66Met variant (
19) was not supported by subsequent longitudinal studies conducted in the context of clinical trials (
20). Furthermore, PRS may have additional advantages in clinical prediction because it is a continuous variable that can have different cutoffs that maximize predictive power, whereas the candidate gene approach can compare only carriers with noncarriers. Moreover, its predictive power will increase as the discovery sample becomes larger.
In the present study, we aimed to investigate whether PRS based on the large-scale GWAS conducted by the Psychiatric Genomics Consortium (
1) was predictive of antipsychotic efficacy in patients with first-episode nonaffective psychosis. There are several advantages of studying first-episode psychosis, such as minimal or no previous drug exposure, increased effect size of genotype-phenotype association (
21), and representation of the whole patient population compared with chronic patient samples that may be subject to ascertainment biases (
22). Although one previous study examined PRS in relation to clinical response to lurasidone in patients with chronic schizophrenia (
23), the present study is the first study, to our knowledge, to longitudinally examine treatment response in patients with first-episode psychosis undergoing initial treatment with antipsychotics.
Discussion
In multiple cohorts of first-episode patients with nonaffective psychosis, we found that schizophrenia PRSs were significantly predictive of antipsychotic drug efficacy, with higher PRSs associated with poorer treatment response. These results suggest that polygenic burden may affect severity of illness, in addition to reflecting risk for developing psychosis. To the best of our knowledge, this is the first study to identify replicable effects of PRS in predicting antipsychotic efficacy in patients undergoing initial treatment for a first episode of illness.
Few previous studies have examined the relationship of PRS to treatment response. Consistent with our findings, a cross-sectional study reported significantly higher PRSs among patients with treatment-resistant schizophrenia (as indexed by clozapine treatment and early, insidious onset and poor premorbid social function) compared with patients who had never been prescribed clozapine (
17). However, in this same study, clozapine responders had higher schizophrenia polygene scores than nonresponders, suggesting that treatment with clozapine may be an important (and perhaps underutilized) treatment option for patients with high PRSs. A second cross-sectional study reported similar results, with clozapine initiation associated with elevated PRS (
18). However, it is noteworthy that results fell short of statistical significance (adjusted hazard ratio=1.23; 95% CI=0.97–1.56), albeit with a smaller sample size (clozapine group, N=105) compared with the previous study (clozapine group, N=434) (
17). It is also noteworthy that the association between PRS and clinical outcome was weaker (and nonsignificant) for more broadly defined treatment resistance based on chart history, indicating the importance of prospective studies (
18). The only longitudinal study to examine the relationship of PRS to treatment response demonstrated a paradoxical inverse relationship, such that higher scores were associated with greater reduction in symptoms after 6 weeks of treatment with lurasidone (
23). It is possible that the ascertainment criteria of this lurasidone clinical trial may have affected results, as patients with treatment-resistant symptoms were explicitly excluded, and patients with good clinical outcomes with standard treatments would not have enrolled in a phase 3 clinical trial of a novel antipsychotic.
Studies of patients in the first episode have the advantage of examining the full range of clinical trajectories of schizophrenia, before patients become lost to research due to either very good or very poor outcomes (
22). Only two reports have examined PRS in the context of first-episode psychosis (
36,
37). In contrast to the present study, both of these studies included patients with affective as well as nonaffective psychosis, but the results were largely consistent with the present findings. The first study revealed higher schizophrenia PRSs among patients ultimately diagnosed with schizophrenia compared with those with affective psychoses (
31). The second study, although longitudinal, did not directly report on treatment-related changes; nevertheless, higher PRS was significantly and positively correlated with PANSS total score after 1 month of treatment (
37).
Pharmacogenetic studies of antipsychotic drug response have typically focused on individual genes and SNPs in the candidate gene approach. Although a few genes have been reported to predict antipsychotic efficacy, such as
DRD2 (
11,
38),
HTR2A (
39,
40), and genes in the glutamate system (
41), most SNPs have had small effect sizes, few have been convincingly replicated (
10,
11), and their clinical significance is questionable. Although dopamine D
2 receptor antagonism is the common, and probably necessary, mechanism of action for antipsychotic drugs, these agents bind to many different receptors of various neurotransmitters (
42), and it is very likely that some of these may be involved in antipsychotic drug response (
43). Perhaps more importantly, many of the drug effects may be from downstream reactions within the dopamine signaling pathway. Therefore, the examination of multiple genes is important because this may help capture the potential downstream effects from antipsychotic drugs. In addition, PRS represents the total genetic burden of liability to schizophrenia. Conceivably, higher genetic burden may implicate a broader range of etiopathophysiologic mechanisms, thereby rendering patients less responsive to drug treatment based primarily on a single mechanism of action (dopaminergic blockade). As such, the PRS approach may be useful in both a practical and a theoretical sense in predicting clinical treatment response.
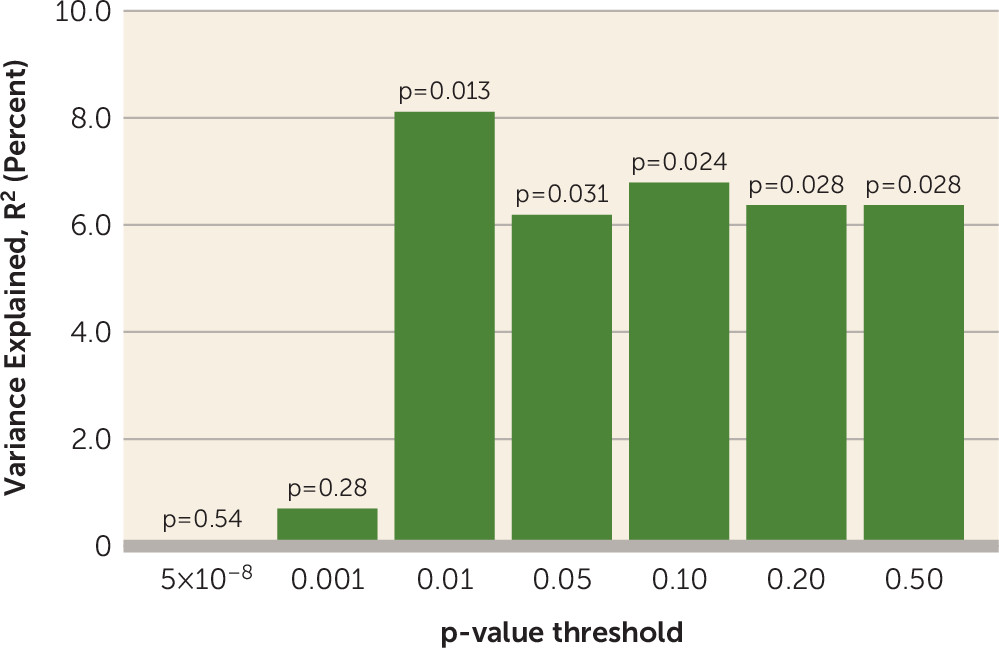
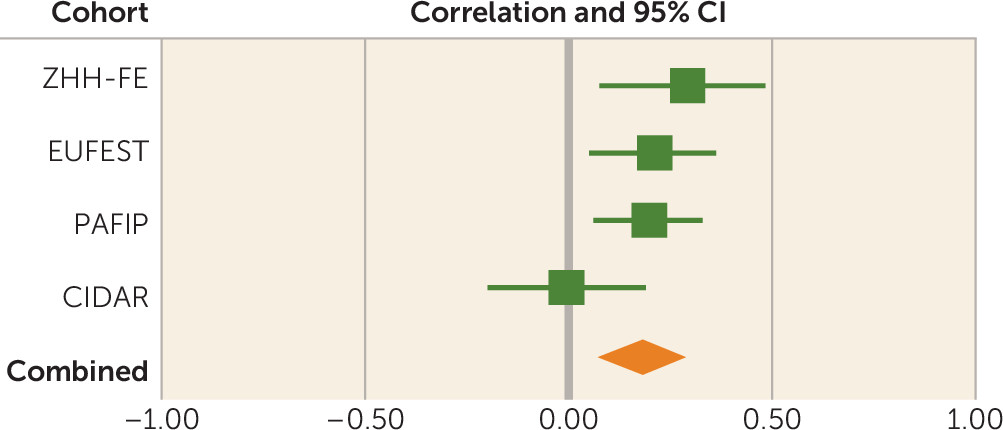
There are several limitations of the present study. PRS is a weighted sum of risk alleles that an individual carries. Many of the SNPs included in PRS may not be relevant to antipsychotic drug response, and inclusion of these could dilute the signal. We observed that statistical association was generally significant by using PRS PT values ≥0.01, suggesting that thousands of SNPs are required in order to saturate the relevant signal, whereas use of only SNPs attaining genome-wide significance in the Psychiatric Genomics Consortium schizophrenia GWAS was insufficient to capture this variance. However, we currently do not have sufficient biological knowledge or statistical techniques to ascertain which SNPs are relevant and which are contributing noise. In addition, the four cohorts of patients examined in the present study were treated with various antipsychotic drugs that could increase the heterogeneity in outcomes, thereby decreasing our ability to detect significant signals. In the future, a very large sample of patients with first-episode psychosis undergoing a single drug treatment would be required to discover which genetic variants are involved in antipsychotic drug response. Finally, PRS was not predictive of antipsychotic treatment response in the CIDAR cohort. If the true effect size is most accurately reflected in the meta-analytic result (r=0.18), then a sample with 100 study subjects (such as the CIDAR cohort) would only have a power of 0.42 to detect a significant effect. Therefore, the failure to replicate in the CIDAR cohort was most likely due to chance variation, possibly exacerbated by the multiethnic nature of the sample. It is noteworthy that the overall pooled effect size was within the 95% confidence interval of the effect size in the CIDAR cohort, and thus this sample is not truly an outlier. At the same time, the effect size observed in the initial discovery cohort (the ZHH-FE sample) was substantially larger than the effect sizes in the remaining cohorts, perhaps reflective of the “winner’s curse.” Given these variable results, it is noteworthy that the meta-analytic effect size (3.24%) was comparable to the effect sizes of the two largest, and most homogeneous, studies (3.5%−3.7%).
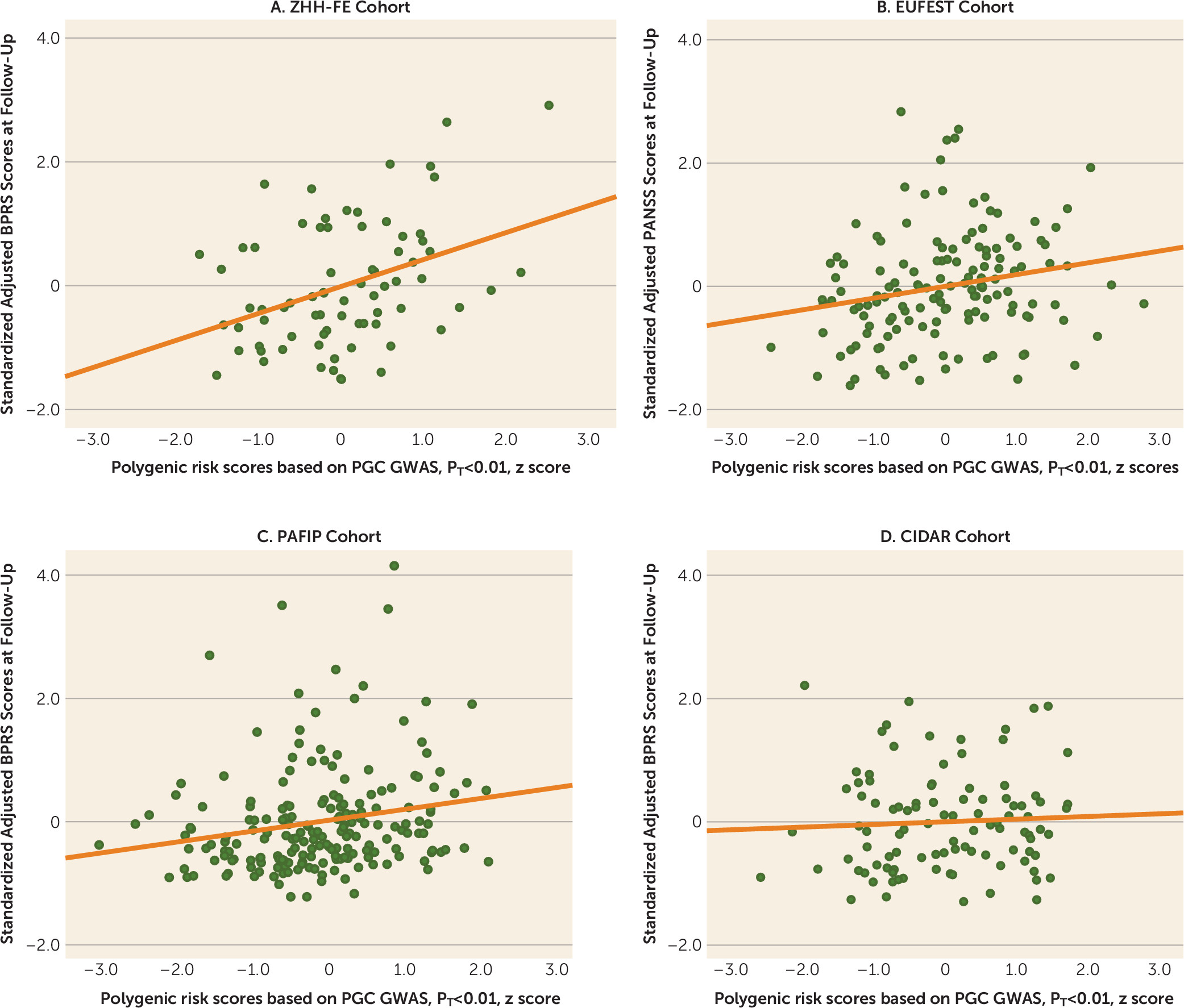
Future studies with larger samples may also result in the ability to identify a PRS cutoff with sufficient explanatory power to attain clinical utility. In the present study, we observed an odds ratio of nearly 2 for dichotomized treatment response among patients with low PRSs compared with patients with high PRSs. Although this effect size is insufficient to guide clinical decision making, a recent large-scale study of PRS in bipolar disorder demonstrated how modest effect sizes may still allow clinical utility at the extremes (
44). With a sample size of 2,586 patients, the International Consortium on Lithium Genetics was able to divide a cohort into deciles on the basis of PRS, whereas the present study was limited to a median split due to relatively smaller sample size. In the International Consortium on Lithium Genetics study, patients with bipolar disorder in the lowest decile of schizophrenia PRS had a nearly 3.5-fold better response rate to lithium compared with patients in the highest decile of PRS. Median split of the International Consortium on Lithium Genetics data would have provided an odds ratio of only 1.68, which is weaker than that observed in the present study. Given the linear relationships observed in the present study (
Figure 2), it is reasonable to hypothesize that a larger sample size could provide an upper cutoff with strong prognostic ability. In this regard, PRS may ultimately be a more flexible and powerful biomarker than individual SNPs, which only permit three genotypic classifications. However, if greater schizophrenia polygenic burden is associated with poorer response to all conventional treatments, enhanced use of clozapine or novel therapeutic approaches (
45) will be even more urgently needed for this subpopulation.