Beyond the glutamate system, ketamine interacts with several additional neurotransmitter systems, including mu, delta, and kappa opioid receptors, and it is currently used as an antinociceptive agent for acute and chronic pain (
8). Ketamine’s analgesic effects are blocked by mu and delta opioid receptor antagonists but not by kappa opioid receptor antagonists, indicating a mu or delta opioid mechanism in ketamine’s antinociceptive effects (
9). We and others have hypothesized that ketamine’s antidepressant mechanism of action may in fact be related to intrinsic opioid receptor properties of ketamine (
10) and have proposed that coadministration of an opioid receptor antagonist with ketamine could be employed to test this hypothesis (
11). Yet, no study to date has probed the role that ketamine’s opioid properties play in its antidepressant effects (
12).
As a dissociative anesthetic (
13,
14), ketamine is capable of producing dramatic psychotomimetic effects (
15–
17), and these effects have been correlated to its antidepressant efficacy (
18). Here too, the specific receptor system or systems responsible for mediating dissociative effects of ketamine are unknown. Some but not all NMDA receptor antagonists cause dissociation (
19). The pure kappa opioid receptor agonist salvinorin A does produce dissociative effects similar to those of ketamine (
20,
21). A low dose (25 mg) of the opioid receptor antagonist naltrexone can augment the psychoactive effects of lower subanesthetic doses (∼0.4 mg/kg per hour) of ketamine, but not higher subanesthetic doses (∼0.6 mg/kg per hour) in healthy subjects (
22). However, opioid receptor antagonists have not been previously used to probe the role opioid receptors play in ketamine’s dissociative effects in adults with treatment-resistant depression, and the 25-mg dose of naltrexone does not completely block opioid receptors (
23).
The objective of this study was not to assess ketamine’s antidepressant efficacy but rather to determine the role of the opioid system in ketamine’s antidepressant and dissociative effects in adults with treatment-resistant depression. We conducted a randomized double-blind crossover trial in which intravenous ketamine was infused once each across two conditions, with participants receiving pretreatment with naltrexone before one of their ketamine infusions (the ketamine plus naltrexone condition) and placebo before the other ketamine infusion (the ketamine plus placebo condition) in a counterbalanced manner. Through this mechanistic clinical trial design, we tested whether pretreatment with an opioid receptor antagonist is able to attenuate the acute antidepressant or dissociative effects of ketamine.
Discussion
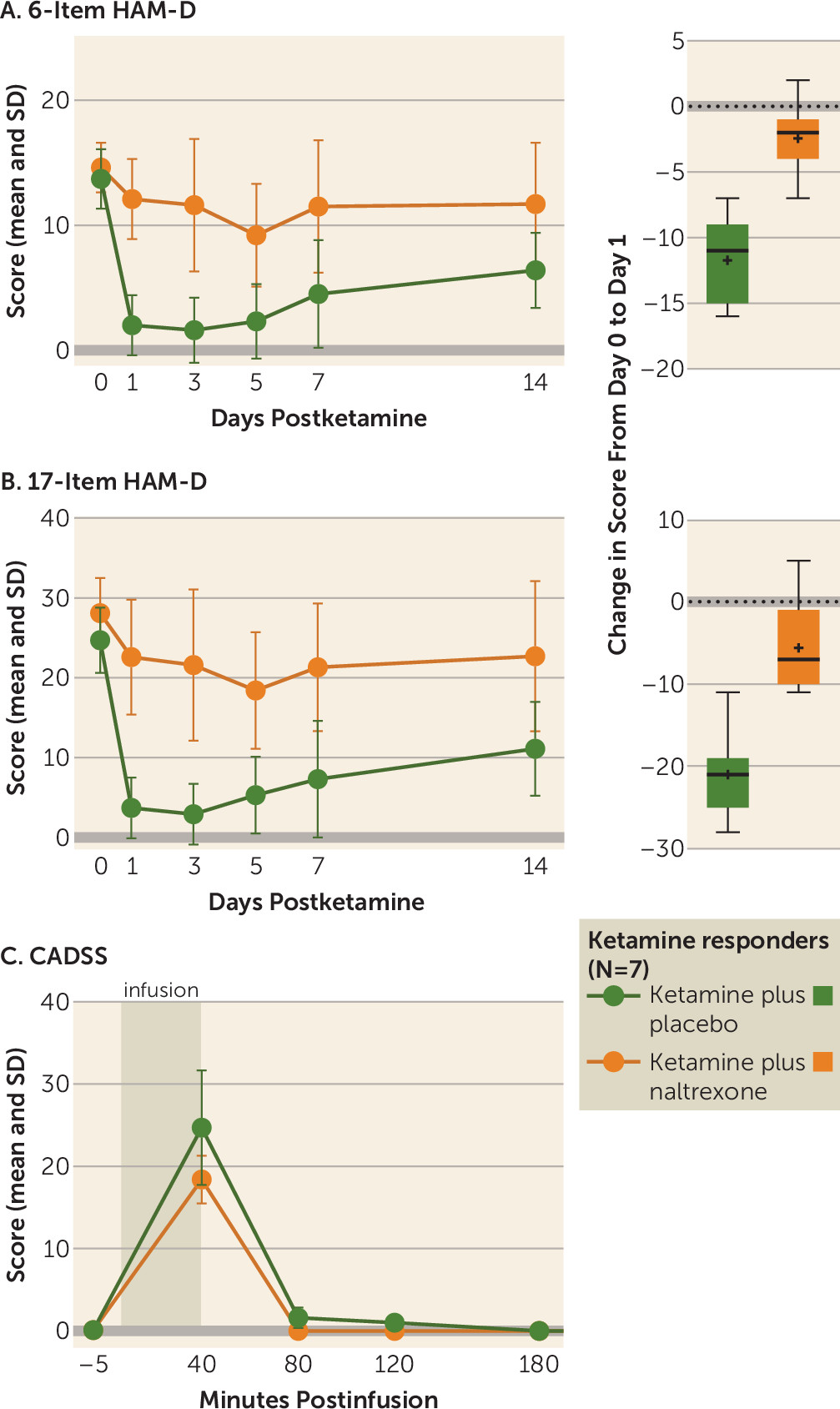
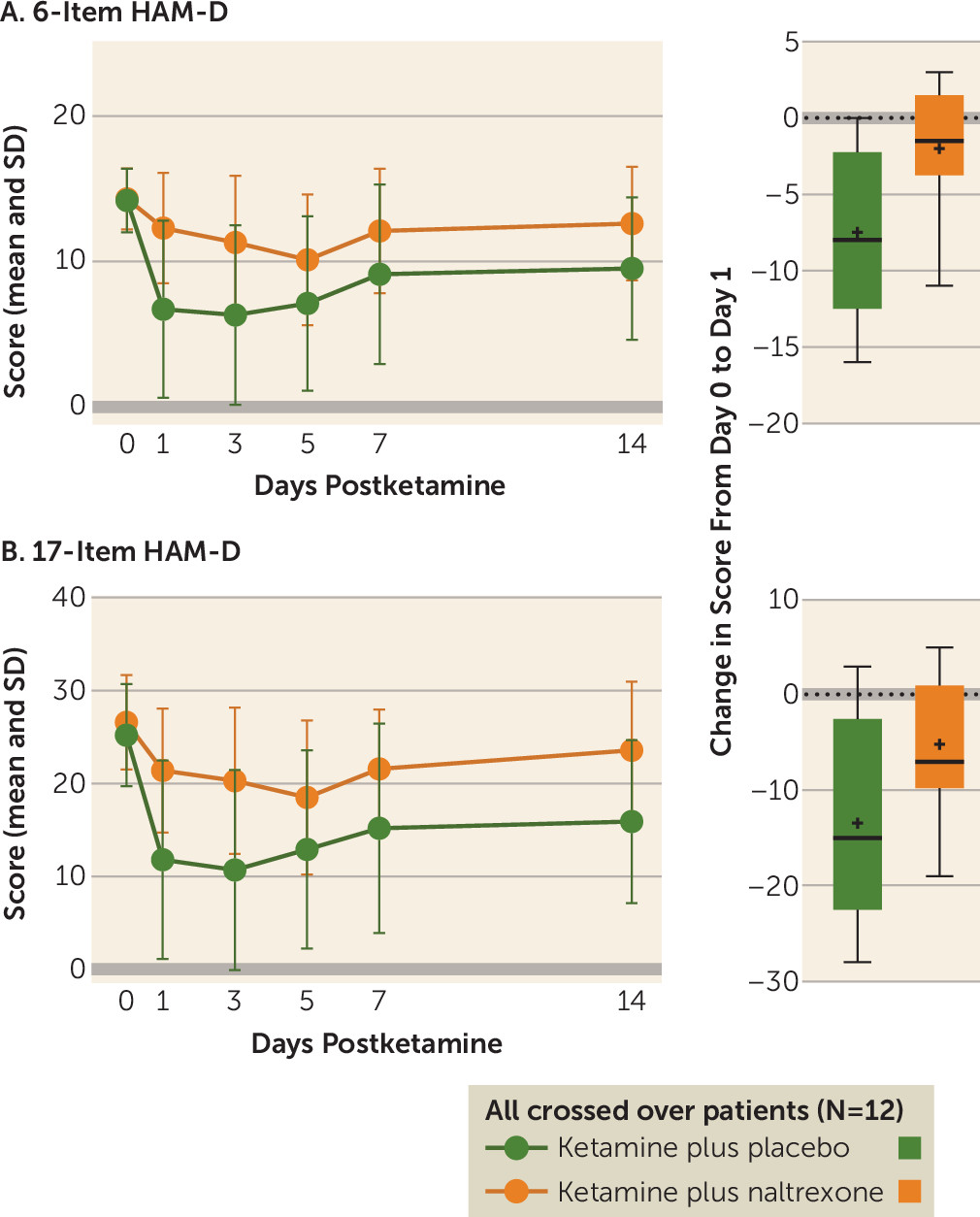
Ketamine has well-established rapid-onset antidepressant effects. The majority of preclinical studies investigating the mechanism of this effect have focused on NMDA receptor antagonism, and several clinical trials have attempted to replicate this rapid antidepressant effect with other NMDA receptor antagonists, with limited success (
5). We now present the first evidence in humans that opioid receptors are necessary for ketamine’s acute antidepressant effect. In ketamine-responsive patients with treatment-resistant depression, pretreatment with naltrexone profoundly attenuated ketamine’s antidepressant effect, with none of the ketamine responders meeting the response criterion at day 1. We observed concordant effects on related measures of depression, including clinician-administered scales (the 6-item HAM-D and the Montgomery-Åsberg Depression Rating Scale [MADRS]) and a self-report instrument (the Beck Depression Inventory–II) (see the
online supplement), which strengthens our conclusion that ketamine’s antidepressant effects require opioid system activation. Of note, we observed a statistically significant difference from baseline at postinfusion day 1 in 17-item HAM-D score for the ketamine plus naltrexone condition, but not on the MADRS or the 6-item HAM-D, both of which are scales thought to reflect core depressive symptoms.
The endogenous opioid system has been reported to play a central role in the pathophysiology and treatment of affective disorders (
38–
43). A robust nonhuman primate literature supports the idea that opioids are important in mediating emotions associated with depression (
44,
45). Depressive disorders have been associated with dysregulation of the endogenous opioid system, particularly mu and kappa opioid receptors’ tone (
39,
40). Moreover, buprenorphine, a mu opioid receptor partial agonist and a kappa opioid receptor antagonist, has been shown to produce antidepressant effects (
41,
42), even in individuals who have failed to benefit from ECT (
43). In obsessive-compulsive disorder, single infusions of ketamine have been reported to produce a multiday benefit (
46), as has a single oral dose of morphine, which is a mu opioid receptor agonist (
47). These data suggest that mu opioid receptor agonists with additional NMDA receptor antagonist properties may have therapeutic potential as intermittently dosed therapies for mood or anxiety disorders.
The kappa opioid receptor is also emerging as a regulator of mood and motivation (
48–
50), with increased kappa opioid receptor activity being associated with depression (
51). Because naltrexone does not have substantial selectivity for the mu opioid receptor over the kappa opioid receptor (
52,
53), the 50-mg dose of naltrexone used in this study saturated the mu opioid receptors and likely equally saturated the kappa receptors (
23,
54). Thus, our data cannot distinguish between the respective roles of mu and kappa opioid receptors in mediating ketamine’s antidepressant effects. Nonetheless, given the available data implicating mu opioid receptor–based mechanisms of antidepressant efficacy, inconsistent findings regarding kappa opioid receptor antagonists in depression (
55,
56), and ketamine’s putative kappa agonist mechanism (
21,
57), we favor the interpretation that ketamine produces its acute antidepressant response primarily through direct and/or indirect actions at the mu opioid receptors. Naltrexone, when chronically administered alone in healthy subjects as well as in individuals with mood and substance use disorders, has been demonstrated across several placebo-controlled trials either to act as an antidepressant or to be mood neutral (
23,
32–
37), which suggests that naltrexone is not simply acting as a depressogenic agent in this case but rather providing selective blockade of the antidepressant effects produced by ketamine.
How do we reconcile these data with the large body of evidence implicating glutamate receptors in ketamine’s primary antidepressant mechanism? The majority of studies to date have focused on ketamine’s antidepressant mechanism of action as a noncompetitive antagonist of the NMDA receptor and subsequent activation of AMPA receptors. Recently, a preclinical study reported that a metabolite of ketamine, 2
R,6
R-hydroxynorketamine, has antidepressant efficacy through stimulation of the AMPA receptor independently of NMDA receptor antagonism (
7). This mechanism of action has been replicated by some (
58,
59) but not all (
60) groups. In addition, preclinical studies demonstrate that glutamate receptor modulation triggers downstream modulation of synthesis and release of brain-derived neurotrophic factor and enhances synaptic plasticity via activation of molecular targets such as mammalian target of rapamycin and eukaryotic elongation factor 2 (
58,
59,
61–
63). These glutamate system effects may in fact drive the transient maintenance of the antidepressant response through modulation of brain plasticity (
64) rather than producing the actual acute antidepressant effects.
No studies to date have directly addressed the role of opioid receptors in ketamine’s antidepressant effect. However, our demonstration of an opioid system activation requirement for ketamine’s acute antidepressant effect mirrors a long-standing literature investigating the opioid mechanism of action of ketamine’s analgesic properties. On the MADRS and the 6-item HAM-D (which reflects the core depressive symptoms [
65]), our data demonstrated that the effect of ketamine was actually ablated by naltrexone. Meta-analyses have consistently shown that ketamine has a clinically significant opioid-sparing effect, in which coadministration of ketamine allows for lower doses of traditional opioids to be used to achieve similar antinociceptive effects (
66). In addition to ketamine’s combined naloxone-sensitive and naloxone-insensitive analgesic effects (
67), human and preclinical studies have found that ketamine 1) substantially potentiates the analgesic effect of opioids (
68), 2) produces opioid receptor–dependent analgesia (
9,
69,
70), 3) reduces opioid tolerance and opioid-induced hyperalgesia to opioids (
71), and 4) produces mu opioid receptor–dependent respiratory depression (
70).
With the proviso that the scope of ketamine’s pharmacology is continually expanding (
72), the available evidence suggests that ketamine-mediated analgesia involves either a direct action at mu opioid receptors (
57,
73–
75) or an interaction between NMDA receptor antagonists and mu opioid receptors (
76–
78). The hypothesis that NMDA receptor antagonists and mu opioid receptors share subcellular co-localization and may exist as a functional complex in a crucial nociceptive brain area (the periaqueductal gray) (
77) forms a particularly compelling explanation for apparently conflicting findings in the context of ketamine-mediated analgesia. Notably, naltrexone pretreatment did not significantly affect ketamine-induced dissociation, as measured by the CADSS, nor did CADSS score correlate with ketamine’s antidepressant efficacy. Previous work has attributed ketamine’s dissociative and hypnotic properties to NMDA receptor antagonism and hyperpolarization-activated cyclic nucleotide-gated cation channel 1 blockade (
72) as well as activation of kappa opioid receptors (
21). Our finding that the dissociative effects of ketamine persist despite naltrexone antagonism of opioid receptors suggests that opioid receptors do not play a major role in mediating ketamine’s dissociative effects.
The public health significance of ketamine’s opioid properties needs to be studied. Depression and opioid dependence are currently the two most significant public health problems facing the United States and have become leading causes of disability and death worldwide (
1,
79,
80). While opioids have a history of use as antidepressants (
43,
81), they pose a significant risk if used chronically (
82). Half of patients who receive prescriptions for opioids have a mental health diagnosis (
50,
83–
85), and over half of individuals with opioid use disorders have a primary diagnosis of depression (
86). There is also a significant ketamine abuse problem worldwide (
87–
89), and ketamine ranks high on the list of commonly abused substances (
90–
94). Moreover, ketamine abusers have high rates of depression (
77) and experience significant brain dysfunction (
95). While these risks have not been demonstrated in serial infusions for depressed patients (
96), short-interval repetitive dosing strategies may pose greater risks (
97), and there have been case reports of apparent tolerance after chronic administration (
98,
99). Ketamine tolerance has been observed in pain/anesthesia indications (
100–
107) as well as in animal models (
108–
110). The route of administration (
111) and the patient’s access to the medication may play a role in the risk (
112). Thus, the abuse and dependence potential of frequent ketamine treatment in major depression needs further study, and our results provide strong justification for further caution against widespread and repeated use of ketamine before further mechanistic testing has been performed (
99,
113,
114).
Our study has a number of strengths and weaknesses. A crossover design was the optimal method for testing the study’s mechanistic hypothesis, since it can clearly identify ketamine responders post hoc and establish, in the individual study participant, that ketamine’s antidepressant effects are mediated via the opioid system. We did not employ an alternative design in which responders would first be identified by open-label treatment with ketamine, which could produce an expectancy bias, with participants expecting that they would have a similar response in the randomized treatment. Moreover, the crossover design provides significantly greater statistical power to detect group differences with fewer subjects. Limitations of a crossover study include potential carryover effects (
115). However, because ketamine’s effects are transient, our washout period was sufficient for participants’ 17-item HAM-D scores to return to within 20% of their baseline scores, and thus any medication-related carryover effects were limited. While we cannot completely rule out the presence of carryover effects in our primary analysis (
115), in an alternative analysis involving only the first randomized infusion (prior to crossover), we did observe a significant difference in response between the ketamine plus placebo and ketamine plus naltrexone conditions (see the
online supplement), further demonstrating that naltrexone blocks the antidepressant effects of ketamine.
We assessed the integrity of the blind post hoc with visual analog scale assessments made 45 minutes after taking naltrexone or placebo, just prior to ketamine infusion. We could identify no item or group of items (see Figure S4 in the
online supplement), or side effect, that participants could have reasonably used to infer their blinded condition. In any longitudinal study, regression to the mean is a possible issue. Data from Murrough et al. (
12) indicate that initial response to ketamine is replicated by reinfusing ketamine three times per week for 2 weeks with repeated treatments. One weakness in our study was the final sample size in the interim analysis, and our findings do need to be replicated in other studies. Still, we found the same qualitative block of ketamine’s effect regardless of the depression instrument used, and with several alternative statistical analyses. We decided to stop the study because our results were both statistically and clinically significant and we were concerned about the ethics of exposing more people to a clearly ineffective and noxious combination treatment.
Further studies are needed to expand our understanding of the opioid effects of ketamine, including studies seeking to determine which opioid receptors are involved in mediating ketamine’s antidepressant effects, using more selective opioid receptor antagonists (
116), surrogate markers (
117), and functional neuroimaging capable of discerning those selective effects (
54). The findings presented here challenge our understanding of the mechanisms of action of ketamine that underlie its potent antidepressant properties (
118,
119).