“A.M.” is a 44-year-old woman who developed treatment-resistant major depressive disorder despite a history of stable and focal bilateral amygdala lesions caused by lipoid proteinosis of Urbach and Wiethe (OMIM #247100), an extremely rare autosomal-recessive disorder resulting from mutations in the extracellular matrix protein 1 gene (ECM1) located on chromosome 1q21 (1). We have repeatedly studied this patient and her monozygotic twin sister, who shares the same Urbach-Wiethe phenotype and amygdala pathology. Brain scans taken longitudinally over the past two decades have confirmed complete destruction of the basolateral amygdala bilaterally (Figure 1A; see also Figure S1 in the online supplement), and a large body of research involving the twins has helped identify brain functions and behaviors that do (2) or do not require amygdala integrity (3). In these studies, A.M. underwent extensive diagnostic screening, which confirmed preserved neuropsychological functioning, including memory, attention, and cognitive flexibility, in the absence of any psychiatric abnormalities, including no history of major depression (4, 5). Perhaps related to the extraordinarily strong, supportive bond with her mother, A.M. (but not her identical twin sister) displayed preserved recognition of fearful faces (6), but emotionally charged pictures never aroused her in a manner similar to the way they did control subjects (7). This was paralleled by a flat affect and the absence of anxiety in life situations experienced as stressful by her family (8).
The onset of major depression for A.M. started 4 years ago, and it was preceded by a series of major life events, all within a 1-year period, and all related to the loss of close attachment figures who were of central importance to her in providing support, care, and connectedness: her mother died from complications from a routine diagnostic medical procedure; she divorced her husband, who left her for another woman; and her teenage son decided to leave home and live with his father. Disabled by grief, broken-heartedness, and exhaustion, she stopped working and moved into her father’s house. She attempted suicide and underwent five inpatient treatments in psychiatric hospitals. Notably, she did not benefit from any of the multimodal treatment attempts, including cognitive-behavioral therapy, multiple classes of antidepressants, and bilateral electroconvulsive therapy (see Table S1 in the online supplement).
On admission to our department last year, A.M. exhibited pronounced symptoms of anhedonia, loss of energy, and pervasive pessimistic thought biases expressing despair, resignation, and a passive wish to die (e.g., “I am hopeless; nothing has ever helped make me feel better; my life won’t ever change for the better”). She displayed sleep disturbances, concentration deficits, and feelings of worthlessness and inferiority. Her cognitive distortions also affected her body image, evident in complaints about her appearance (e.g., “I am so ugly; I am not attractive to anybody; I will never find another partner who wants to share his life with me”). She refused to wear glasses despite moderate myopia. Her loss of appetite resulted in a diagnosis of cachexia (wasting syndrome) on admission (body mass index, 17.75). Furthermore, she suffered from a dry mouth, salty taste, and painful oropharyngeal ulcerations typical of Urbach-Wiethe disease.
Compared with a premorbid assessment 7 years earlier (6), her social network had declined both in size (the number of people in her social network was 17 in 2012, and 10 in 2018) and complexity (four embedded networks in 2012, and one in 2018; see the online supplement). In addition, she exhibited high levels of loneliness (a UCLA Loneliness Scale score of 47; see the online supplement). Clinical interviews confirmed that she had no other psychiatric disorder than major depressive disorder. Ten years ago, in an exploratory positron emission tomography study using the radioligand [18F]altanserin (5), we detected a significantly decreased expression of serotonin (5-HT2A) receptors throughout A.M.’s brain, which may be linked to her amygdala pathology in ways neither investigated nor understood.
In light of the treatment-refractory and suicidal nature of A.M.’s major depression, the rationale of our therapeutic strategy was rooted in the assumption that her condition may be nonresponsive to serotonin-based therapies and may instead benefit from intravenous ketamine, which has shown transient efficacy for treatment-resistant symptoms in suicidal depression (9). Given associations that have been identified between major depression and dysfunction in large-scale brain networks (see the main text, below), we also decided to probe the effects of ketamine on the default mode network (DMN), salience network (SAN), and frontoparietal network (FPN) in the patient by collecting a series of resting-state functional MRI (rsfMRI) scans before and after her transient recovery from depression.
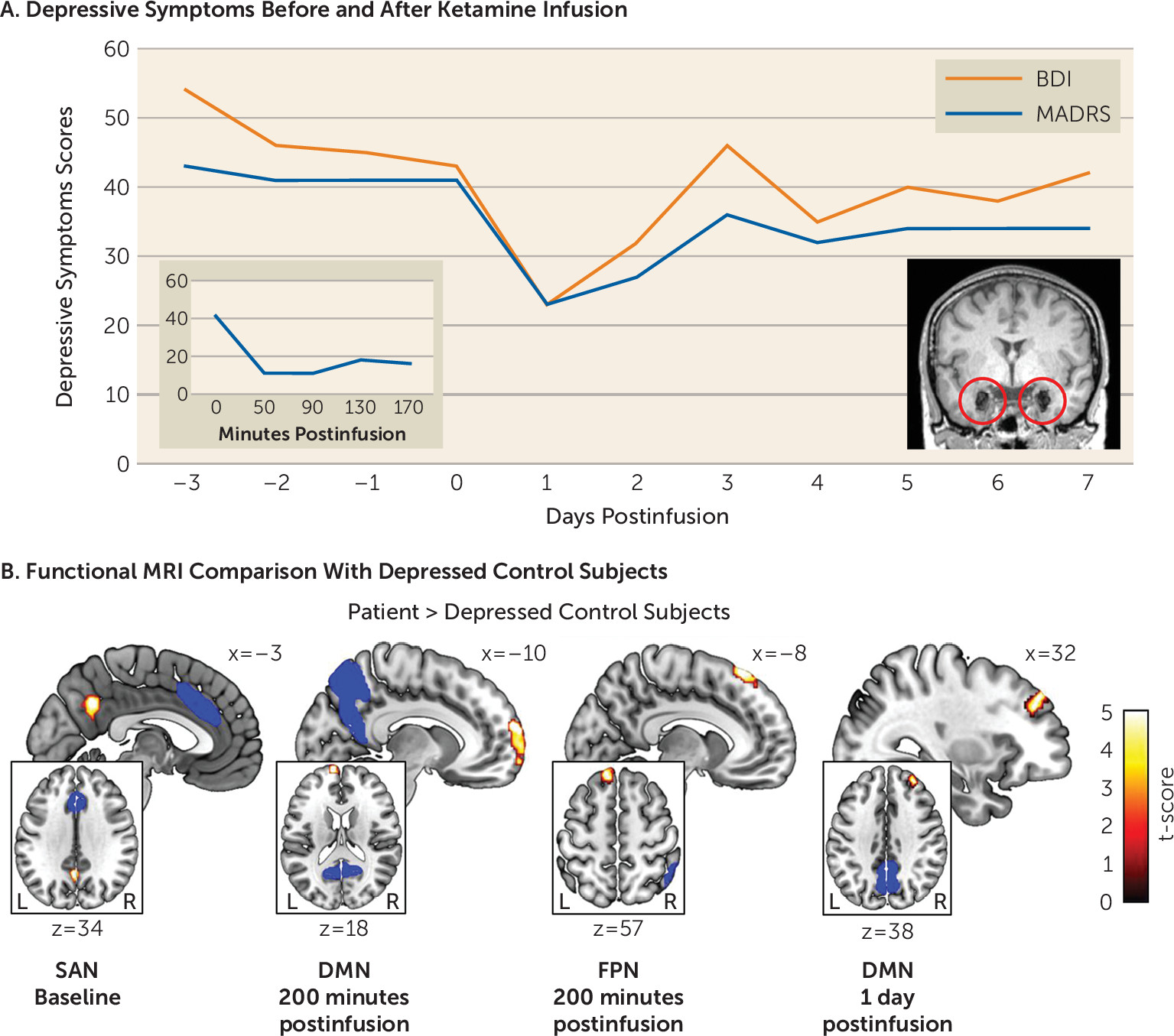
Four days after the patient was admitted to our hospital, intravenous ketamine (0.5 mg/kg) was administered over 40 minutes. The patient underwent rsfMRI scanning at baseline on admission as well as 200 minutes, 1 day, and 7 days after ketamine infusion. The rsfMRI data were compared with 12 sex- and age-matched inpatients with major depression who underwent scanning only once, at baseline; a voxel-based morphometry analysis showed normal amygdala gray matter volume in these patients (see the online supplement). Depressive symptoms were measured with the Beck Depression Inventory–II (BDI), the Montgomery-Åsberg Depression Rating Scale (MADRS), and the Hamilton Depression Rating Scale (see the online supplement). Suicidal ideation was assessed with the Columbia-Suicide Severity Rating Scale (C-SSRS). Dissociative symptoms were evaluated with the Clinician-Administered Dissociative States Scale (CADSS).
On admission, the patient suffered from severe depression (scores of 43 on the MADRS, 54 on the BDI, and 21 on the C-SSRS). Antidepressant response was evident 50 minutes after ketamine infusion (a 73% reduction in MADRS score; the patient also had a 65% reduction in BDI score 170 minutes after infusion; see Figure 1A), with depressive symptoms returning to baseline after 3 days. In stark contrast to her previous behavior, she started showing interest in the lives of other patients and caretakers and proactively initiated conversations (e.g., “What are your plans for the holidays?”). The intensity of her suicidal ideation also substantially dropped (a score of 13 on the C-SSRS 200 minutes after infusion). Together, these observations suggest that ketamine transiently disrupted her negative self-referential bias and suicidal depression. Ketamine also induced dissociative symptoms that peaked 50 minutes after the infusion (a score of 24 on the CADSS; e.g., “I cannot feel my body, it’s like being weightless”) and vanished after 130 minutes (a score of 2 on the CADSS).
Results from rsfMRI analysis showed significant differences between the patient’s measurements and those of the depression control group in the DMN, FPN, and SAN. Specifically, at baseline the patient exhibited significantly increased functional connectivity between the medial prefrontal cortex as seed of the DMN and the superior frontal gyrus (peak Montreal Neurological Institute coordinates: 12, −6, 76; t=4.02 [df=11 throughout], false discovery rate-corrected p [pFDR]<0.01), and the anterior cingulate cortex as seed of the SAN and the precuneus (−4, −56, 34; t=4.02, pFDR<0.01). Both differences were no longer detectable 200 minutes and 1 day after ketamine infusion, but the altered SAN-precuneus connectivity was again evident after 7 days (−2, −60, 32; t=5.12, pFDR=0.06). During the acute period after ketamine infusion, across DMN seed regions, we found significantly increased coupling between the DMN and the frontal pole (left lateral parietal cortex as seed: −12, 66, 4; t=8.95, pFDR<0.01; medial prefrontal cortex as seed: −8, 70, 2; t=6.92, pFDR<0.01; posterior cingulate cortex as seed: −10, 64, 18; t=10.22, pFDR<0.01) (Figure 1B) in the patient 200 minutes after infusion and less pronounced after 1 day (medial prefrontal cortex seed: 30, 40, 34; t=5.65, pFDR=0.08; posterior cingulate cortex seed: 32, 44, 38; t=6.78, pFDR<0.01) compared with the depression control group. Likewise, we observed significantly increased functional connectivity between the right posterior parietal cortex as seed of the FPN and the frontal pole 200 minutes after infusion (−12, 38, 58; t=8.10, pFDR<0.01).
FIGURE 1. Depressive symptoms before and after ketamine administration in a patient with bilateral amygdala damage and severe treatment-resistant depression, and functional MRI comparison with depressed control subjectsa
a Depressive symptoms were assessed for 10 consecutive days after the first subanesthetic intravenous dose (0.5 mg/kg) of ketamine in a patient who had severe treatment-resistant depression despite bilateral basolateral amygdala damage (panel A). Both self-reported and clinician-evaluated depressive symptoms were reduced after ketamine administration. The two inlays display changes in the clinician-rated depressive symptoms in the 170 minutes after the ketamine infusion on the left and a high-resolution sagittal anatomical T1-weighted MR image of the patient’s brain with circles indexing the focal bilateral amygdala calcification damage on the right. Functional MRI was used to measure the default mode network (DMN), frontoparietal network (FPN), and salience network (SAN) at baseline (i.e., 3 days before the infusion) and 200 minutes, 1 day, and 7 days after infusion (panel B). Compared with 12 depressed control subjects (mean age=43 years, SD=13.2), at baseline the patient showed significantly increased functional connectivity between the anterior cingulate cortex as seed of the SAN and the precuneus (−4, −56, 34; t=4.02 [df=11 throughout], pFDR<0.01). Two hundred minutes after the ketamine infusion, the patient exhibited significantly increased functional coupling between the frontal pole and the posterior cingulate cortex as seed of the DMN (−10, 64, 18; t=10.22, pFDR<0.01) and the posterior parietal cortex as seed of the FPN (−12, 38, 58; t=8.10, pFDR<0.01) compared with the depressed controls. Significantly increased functional connectivity between the frontal pole and the posterior parietal cortex as seed of the DMN was also evident one day after the infusion (32, 44, 38; t=6.78, pFDR<0.01). The blue areas illustrate the seed regions. BDI=Beck Depression Inventory–II; MADRS=Montgomery-Åsberg Depression Rating Scale.
To our knowledge, this is the first reported case demonstrating that both treatment-resistant major depression and its response to ketamine can occur in the absence of the basolateral amygdala. Influential reports regarding the neurobiological origin of depression have highlighted a central role for the amygdala in the pathogenesis of depression (10–12). While there are heterogeneous findings of amygdala activity in major depression, possibly as a result of differences in contrast selection (13, 14), recent meta-analytic evidence indicates that major depression is associated with blunted amygdala responses to negative stimuli (13). Likewise, a meta-analysis found reduced amygdala volume in unmedicated patients (15), but there are also reports of amygdala enlargement in acutely depressed patients (16). Notably, pretreatment amygdala hyporeactivity has been identified as a general predictor of treatment response (17), and neurofeedback-based increases in amygdala hemodynamic activity can mitigate depressive symptoms (18). Furthermore, major depression has also been associated with dysfunctions in large-scale brain networks (19, 20). One of the most consistent findings is hyperconnectivity of the default mode network (DMN), which encompasses the posterior and anterior cortical midline structures and shows increased activation during self-referential processing in the resting state (21). It has been suggested that the DMN assigns valence to internally represented stimuli, and the DMN has been linked to self-focused rumination in major depression (22). By contrast, patients with major depression have been found to exhibit hypoconnectivity within the frontoparietal network (FPN) and salience network (SAN). The FPN plays a pivotal role in cognitive control of emotional responses (23) and the SAN, comprising the dorsal anterior cingulate cortex, fronto-insular cortex, and amygdala, is crucially involved in determining the biological significance of external stimuli (24). Interestingly, multiple depressive episodes may lead to a temporal decoupling of the amygdala from SAN regions (25).
Although we are not aware of any past reports of treatment-resistant major depression following bilateral amygdala damage, there have been reports of depressive symptoms in amygdala-lesioned patients, such as in the original case study of patient S.M. (32), where it was noted that “she has occasionally reported depressive symptomatology, related to difficult situational exigencies.” A more recent report (33) confirmed this observation and noted that one of the most difficult situations for patient S.M. is her social isolation, leading to feelings of loneliness and abandonment. Similarly, A.M. only developed major depression after a series of adverse life events, all happening in quick succession, and all involving the loss of close family members and feelings of loneliness and abandonment. If A.M.’s twin sister were to experience a similar fate, it remains possible that she would also be at risk for developing major depression. Clearly, a bilateral amygdala lesion is not sufficient for the development of major depression, but it may render individuals more vulnerable to the effects of social isolation, which appears to be a common consequence of having amygdala damage in free-ranging rhesus monkeys (34).
A.M.’s depression featured pronounced and uncontrollable negative cognitive biases and ruminations, and it is possible that an intact amygdala normally helps to inhibit such dysfunctional thought processes, although this is still speculative and needs to be explored further. It has been hypothesized that the amygdala updates the valuation of “self” representations in the orbital frontal cortex (OFC) (35), and we recently showed (36) that an intact amygdala is required to protect us from illusory body experiences and distortions in self-perception. Interestingly, lesions of the basolateral amygdala hinder the formation of stimulus-outcome representations in the OFC of rats (37) and amygdala lesions in macaques significantly reduced, but did not abolish, the encoding of reward value in the OFC (38). Amygdala lesions in humans have also been found to result in reduced OFC activation associated with reward expectation (39). In the present study, at baseline, A.M. exhibited increased connectivity between the SAN and the precuneus, a functional core of the DMN. It has been found that structural integrity of the SAN is necessary for the efficient regulation of activity in the DMN (40). Thus, amygdala damage may affect the homeostatic interplay between large-scale networks (20), possibly facilitating hyperconnectivity within the DMN and leading to the self-centered, ruminative responding characteristic of major depression.
The recent discovery of rapid antidepressant effects of ketamine has stimulated a reconceptualization of how treatment-resistant major depression and suicidality could be targeted, but the mechanisms of action of ketamine remain obscure (26). Ketamine infusions in patients with treatment-resistant major depression have been found to induce an increase in glucose metabolism in the prefrontal cortex that correlates with the opposite effect in the amygdala (27). These changes could be causally involved in the antidepressant effect or a by-product of the symptom reduction. Interestingly, in mouse models of depression, infusion of ketamine into the amygdala was found to have no effects (28), while a subanesthetic intraperitoneal dose of ketamine normalized depressive-like behavior and was accompanied by reduced glutamate functional connectivity strength (29). In patients with major depression, ketamine was found to normalize insular connectivity with the DMN (30) and to increase global connectivity in the prefrontal cortex (31).
In the case of A.M., ketamine was able to initiate a rapid antidepressant effect that was associated with a reduction in connectivity between the DMN and SAN, and an enhancement of connectivity between the DMN and FPN (Figure 1). However, these findings should be interpreted cautiously given the limitations of an open-label case study and A.M.’s unique depression phenotype and brain lesion. While A.M. has complete destruction of the basolateral amygdala, we cannot rule out the possibility that functional residual tissue in the central amygdala and the amygdalo-hippocampal transition zone or damage to fibers passing through the calcified regions contributed to the observed results. Furthermore, it is conceivable that ketamine’s effect on functional connectivity was altered by A.M.’s amygdala pathology, making the fMRI findings highly specific to A.M.’s brain.
The case of A.M. illustrates that treatment-resistant major depression can develop despite focal bilateral amygdala damage, highlighting the fact that the amygdala is not necessary for the subjective experience or behavioral presentation of clinical depression. Current conceptions of major depression emphasize heterogeneity in clinical phenotypes (41) and underlying biotypes (42–44), such that amygdala-based biomarkers may prove insightful for some but not all subtypes of the illness. Moreover, consistent with A.M.’s amygdala lesion, her major depression phenotype was characterized by marked anhedonia and cognitive biases but only modest symptoms of anxiety (45). Given the broad spectrum of major depression phenotypes, it is conceivable that the antidepressant effect of ketamine in subgroups of patients with major depression with strong anxiety features or comorbidities may act via amygdala-dependent mechanisms, but our observations show that ketamine can rapidly exert its antidepressant effects with or without the amygdala.
Acknowledgments
The authors thank Petra Broich, M.D., and Michael Trauscheid, M.D., for their generous support in inpatient treatment. They also thank the Brain and Behavior Research Foundation for a NARSAD Young Investigator Grant (to Dr. Feinstein). Dr. Scheele and Dr. Hurlemann are supported by a German Research Foundation grant (SCHE 1913/5-1 and HU 1302/11-1), a German-Israel Foundation for Scientific Research and Development grant (I-1428-105.4/2017), and an Else-Kröner-Fresenius-Stiftung grant (2017_A35).
Hamada T, McLean WH, Ramsay M, et al: Lipoid proteinosis maps to 1q21 and is caused by mutations in the extracellular matrix protein 1 gene (ECM1). Hum Mol Genet 2002; 11:833–840
Wang S, Yu R, Tyszka JM, et al: The human amygdala parametrically encodes the intensity of specific facial emotions and their categorical ambiguity. Nat Commun 2017; 8:14821
Patin A, Hurlemann R: Behavioral consequences and compensatory adaptations after early bilateral amygdala damage in monozygotic twins, in Living Without an Amygdala. Edited by Amaral DG, Adolphs R. New York, Guilford, 2016, pp 306–333
Zarate CA Jr, Singh JB, Carlson PJ, et al: A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 2006; 63:856–864
Schulze L, Schulze A, Renneberg B, et al: Neural correlates of affective disturbances: a comparative meta-analysis of negative affect processing in borderline personality disorder, major depressive disorder, and posttraumatic stress disorder. Biol Psychiatry Cogn Neurosci Neuroimaging 2019; 4:220–232
Hamilton JP, Etkin A, Furman DJ, et al: Functional neuroimaging of major depressive disorder: a meta-analysis and new integration of base line activation and neural response data. Am J Psychiatry 2012; 169:693–703
Hamilton JP, Siemer M, Gotlib IH: Amygdala volume in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Mol Psychiatry 2008; 13:993–1000
van Eijndhoven P, van Wingen G, van Oijen K, et al: Amygdala volume marks the acute state in the early course of depression. Biol Psychiatry 2009; 65:812–818
Williams LM, Korgaonkar MS, Song YC, et al: Amygdala reactivity to emotional faces in the prediction of general and medication-specific responses to antidepressant treatment in the randomized iSPOT-D trial. Neuropsychopharmacology 2015; 40:2398–2408
Young KD, Siegle GJ, Zotev V, et al: Randomized clinical trial of real-time fMRI amygdala neurofeedback for major depressive disorder: effects on symptoms and autobiographical memory recall. Am J Psychiatry 2017; 174:748–755
Kaiser RH, Andrews-Hanna JR, Wager TD, et al: Large-scale network dysfunction in major depressive disorder: a meta-analysis of resting-state functional connectivity. JAMA Psychiatry 2015; 72:603–611
Fox MD, Snyder AZ, Vincent JL, et al: The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc Natl Acad Sci USA 2005; 102:9673–9678
Hamilton JP, Farmer M, Fogelman P, et al: Depressive rumination, the default-mode network, and the dark matter of clinical neuroscience. Biol Psychiatry 2015; 78:224–230
Marek S, Dosenbach NUF: The frontoparietal network: function, electrophysiology, and importance of individual precision mapping. Dialogues Clin Neurosci 2018; 20:133–140
Jacobs RH, Barba A, Gowins JR, et al: Decoupling of the amygdala to other salience network regions in adolescent-onset recurrent major depressive disorder. Psychol Med 2016; 46:1055–1067
Li CT, Chen MH, Lin WC, et al: The effects of low-dose ketamine on the prefrontal cortex and amygdala in treatment-resistant depression: a randomized controlled study. Hum Brain Mapp 2016; 37:1080–1090
Shirayama Y, Hashimoto K: Effects of a single bilateral infusion of R-ketamine in the rat brain regions of a learned helplessness model of depression. Eur Arch Psychiatry Clin Neurosci 2017; 267:177–182
McGirr A, LeDue J, Chan AW, et al: Cortical functional hyperconnectivity in a mouse model of depression and selective network effects of ketamine. Brain 2017; 140:2210–2225
Evans JW, Szczepanik J, Brutsché N, et al: Default mode connectivity in major depressive disorder measured up to 10 days after ketamine administration. Biol Psychiatry 2018; 84:582–590
Abdallah CG, Averill LA, Collins KA, et al: Ketamine treatment and global brain connectivity in major depression. Neuropsychopharmacology 2017; 42:1210–1219
Feinstein JS, Adolphs R, Tranel D: A tale of survival from the world of Patient SM, in Living Without an Amygdala. Edited by Amaral DG, Adolphs R. New York, Guilford, 2016, pp 1–38
Schoenbaum G, Setlow B, Saddoris MP, et al: Encoding predicted outcome and acquired value in orbitofrontal cortex during cue sampling depends upon input from basolateral amygdala. Neuron 2003; 39:855–867
Rudebeck PH, Mitz AR, Chacko RV, et al: Effects of amygdala lesions on reward-value coding in orbital and medial prefrontal cortex. Neuron 2013; 80:1519–1531
Hampton AN, Adolphs R, Tyszka MJ, et al: Contributions of the amygdala to reward expectancy and choice signals in human prefrontal cortex. Neuron 2007; 55:545–555
Fried EI, Nesse RM: Depression is not a consistent syndrome: an investigation of unique symptom patterns in the STAR*D study. J Affect Disord 2015; 172:96–102
Simmons WK, Burrows K, Avery JA, et al: Depression-related increases and decreases in appetite: dissociable patterns of aberrant activity in reward and interoceptive neurocircuitry. Am J Psychiatry 2016; 173:418–428
Kalin NH, Shelton SE, Davidson RJ: The role of the central nucleus of the amygdala in mediating fear and anxiety in the primate. J Neurosci 2004; 24:5506–5515
Division of Medical Psychology (Scheele, Zimbal, Mielacher, Hurlemann), Department of Anesthesiology (Delis, Neumann), and Department of Psychiatry (Philipsen, Hurlemann), University Hospital, Bonn, Germany; Laureate Institute for Brain Research, Tulsa, Okla. (Feinstein); and Department of Psychiatry, University of Oldenburg Medical Campus, Bad Zwischenahn, Germany (Hurlemann).
Division of Medical Psychology (Scheele, Zimbal, Mielacher, Hurlemann), Department of Anesthesiology (Delis, Neumann), and Department of Psychiatry (Philipsen, Hurlemann), University Hospital, Bonn, Germany; Laureate Institute for Brain Research, Tulsa, Okla. (Feinstein); and Department of Psychiatry, University of Oldenburg Medical Campus, Bad Zwischenahn, Germany (Hurlemann).
Division of Medical Psychology (Scheele, Zimbal, Mielacher, Hurlemann), Department of Anesthesiology (Delis, Neumann), and Department of Psychiatry (Philipsen, Hurlemann), University Hospital, Bonn, Germany; Laureate Institute for Brain Research, Tulsa, Okla. (Feinstein); and Department of Psychiatry, University of Oldenburg Medical Campus, Bad Zwischenahn, Germany (Hurlemann).
Division of Medical Psychology (Scheele, Zimbal, Mielacher, Hurlemann), Department of Anesthesiology (Delis, Neumann), and Department of Psychiatry (Philipsen, Hurlemann), University Hospital, Bonn, Germany; Laureate Institute for Brain Research, Tulsa, Okla. (Feinstein); and Department of Psychiatry, University of Oldenburg Medical Campus, Bad Zwischenahn, Germany (Hurlemann).
Division of Medical Psychology (Scheele, Zimbal, Mielacher, Hurlemann), Department of Anesthesiology (Delis, Neumann), and Department of Psychiatry (Philipsen, Hurlemann), University Hospital, Bonn, Germany; Laureate Institute for Brain Research, Tulsa, Okla. (Feinstein); and Department of Psychiatry, University of Oldenburg Medical Campus, Bad Zwischenahn, Germany (Hurlemann).
Division of Medical Psychology (Scheele, Zimbal, Mielacher, Hurlemann), Department of Anesthesiology (Delis, Neumann), and Department of Psychiatry (Philipsen, Hurlemann), University Hospital, Bonn, Germany; Laureate Institute for Brain Research, Tulsa, Okla. (Feinstein); and Department of Psychiatry, University of Oldenburg Medical Campus, Bad Zwischenahn, Germany (Hurlemann).
Division of Medical Psychology (Scheele, Zimbal, Mielacher, Hurlemann), Department of Anesthesiology (Delis, Neumann), and Department of Psychiatry (Philipsen, Hurlemann), University Hospital, Bonn, Germany; Laureate Institute for Brain Research, Tulsa, Okla. (Feinstein); and Department of Psychiatry, University of Oldenburg Medical Campus, Bad Zwischenahn, Germany (Hurlemann).
Division of Medical Psychology (Scheele, Zimbal, Mielacher, Hurlemann), Department of Anesthesiology (Delis, Neumann), and Department of Psychiatry (Philipsen, Hurlemann), University Hospital, Bonn, Germany; Laureate Institute for Brain Research, Tulsa, Okla. (Feinstein); and Department of Psychiatry, University of Oldenburg Medical Campus, Bad Zwischenahn, Germany (Hurlemann).
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing PsychiatryOnline@psych.org or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).
If the address matches an existing account you will receive an email with instructions to retrieve your username
Create a new account
Change Password
Password Changed Successfully
Your password has been changed
Login
Reset password
Can't sign in? Forgot your password?
Enter your email address below and we will send you the reset instructions
If the address matches an existing account you will receive an email with instructions to reset your password.
Change Password
Congrats!
Your Phone has been verified
×
As described within the American Psychiatric Association (APA)'s Privacy Policy and Terms of Use, this website utilizes cookies, including for the purpose of offering an optimal online experience and services tailored to your preferences. Please read the entire Privacy Policy and Terms of Use. By closing this message, browsing this website, continuing the navigation, or otherwise continuing to use the APA's websites, you confirm that you understand and accept the terms of the Privacy Policy and Terms of Use, including the utilization of cookies.