Schizophrenia and bipolar disorder share some genetic (
1) and environmental (
2) risk factors, and for some individuals, certain clinical features such as psychosis (
3). These shared attributes across diagnoses may be linked via similar functional (
4), morphological (
5), or molecular (
6) brain alterations, many of which have been associated with evidence of mitochondrial dysfunction (
7).
Mitochondria are responsible for multiple processes essential for brain function (
8,
9). The primary role of mitochondria is energy production via synthesis of adenosine triphosphate (ATP) by oxidative phosphorylation (OXPHOS). In neurons, most of the ATP generated supports cell firing and synaptic neurotransmission (
10). Mitochondria also participate in other key functional pathways, including reactive oxygen species generation, Ca
2+ buffering, and apoptosis (
11). Accumulating evidence implicates alterations in one or more of these mitochondrial functional pathways in schizophrenia (
12) and bipolar disorder (
13). Determining the presence, severity, and functional pathway specificity of mitochondrial alterations in schizophrenia and bipolar disorder at multiple anatomical levels of resolution can inform us on the contributors to, and possible therapeutic targets for, neuronal dysfunction in schizophrenia and bipolar disorder.
In vivo indices of cortical neuronal activity and energy metabolism indicate less neuronal OXPHOS in schizophrenia (
14,
15), findings supported by lower transcriptomic, proteomic, and enzymatic measures of energy production in postmortem brain from schizophrenia subjects (
6,
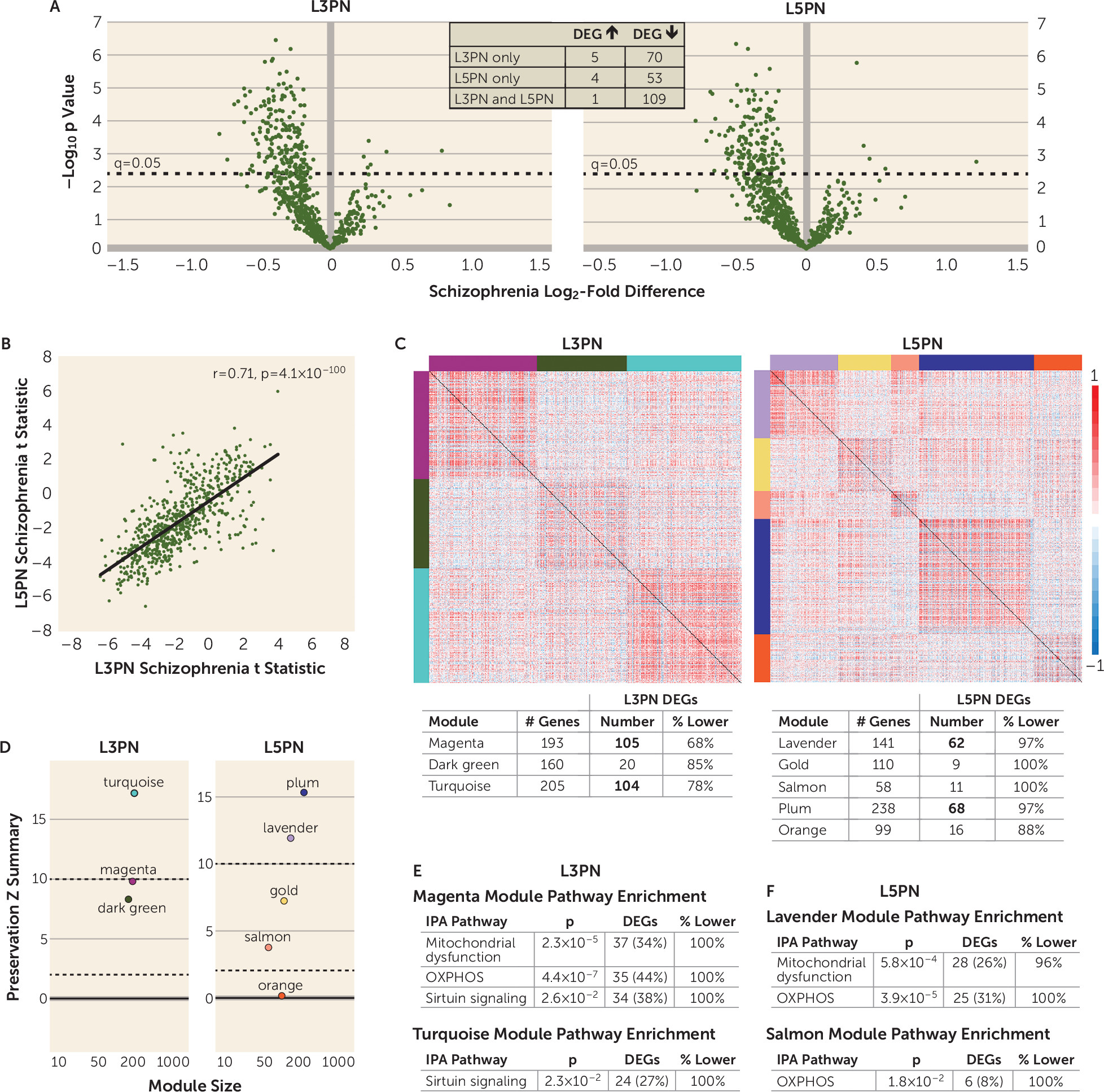
16). Transcriptomic profiling of two key circuit components in the dorsolateral prefrontal cortex (DLPFC), layer 3 pyramidal neurons (L3PNs) and layer 5 pyramidal neurons (L5PNs), revealed that the top affected functional pathways in schizophrenia were related to mitochondrial energy production (
17–
19). In bipolar disorder subjects, in vivo and postmortem findings regarding mitochondrial-related alterations from gray matter (
6,
14,
20–
23) and cell type-specific (
18) studies are mixed. However, a meta-analysis identified oxidative damage to lipids in bipolar disorder subjects (
24), suggesting altered reactive oxygen species regulation.
Together, these data suggest that cortical mitochondrial perturbations are present in both schizophrenia and bipolar disorder but that the severity of alterations and/or the affected mitochondrial functional pathways may differ across diagnoses, with such differences influenced by the anatomical resolution of analysis. To address this issue, we analyzed in schizophrenia and bipolar disorder subjects transcriptomic data that index mitochondrial functional pathways in samples of DLPFC total gray matter, L3PNs, and L5PNs using a dual strategy. First, we identified differentially expressed genes (DEGs) and assessed their enrichment for functional pathways, and second, we applied weighted gene coexpression network analysis for an unbiased examination of how disease-related differences in gene expression affect higher-order gene coexpression relationships.
Discussion
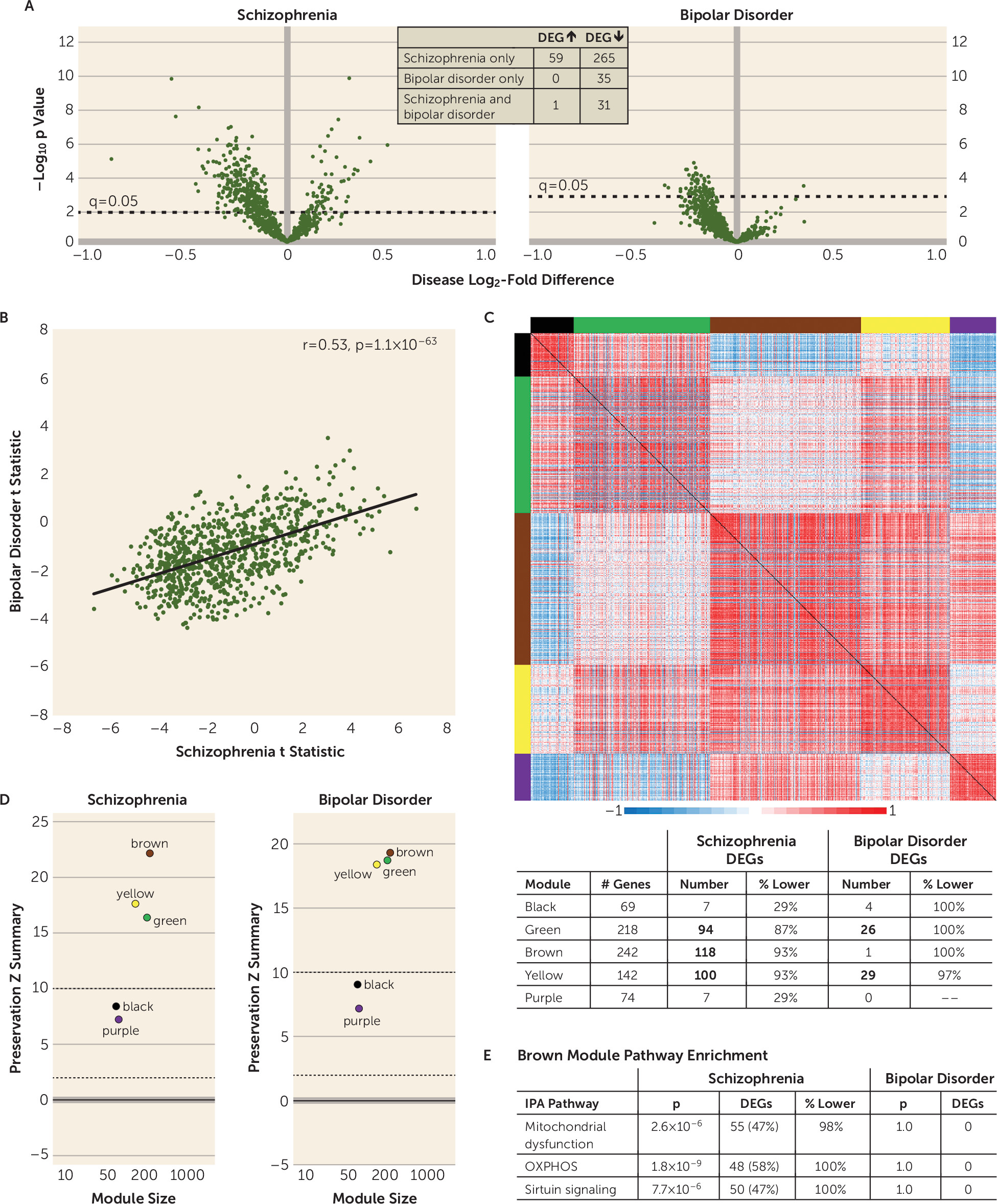
Previous studies of postmortem human cortex suggested that there are mitochondrial perturbations in individuals with schizophrenia and bipolar disorder, but whether the severity of these alterations and/or the affected mitochondrial functional pathways differed across diagnoses was unclear. In this targeted analysis of mitochondrial-related gene expression, we found alterations in total DLPFC gray matter in both schizophrenia and bipolar disorder subjects, but the disease effect was substantially stronger in schizophrenia. Additionally, alterations in mitochondrial functional pathways, all of which were related to lower energy production, were found only in schizophrenia subjects. In pyramidal neurons, mitochondrial-related alterations were exclusively present in schizophrenia subjects and were also enriched for functional pathways indexing lower energy production. Together, these findings support the idea that mitochondrial perturbations are present in the DLPFC in both schizophrenia and bipolar disorder, but that the severity and nature of these alterations, and their apparent cell type-specificity, differ across diagnoses.
In the analyses of mitochondrial-related genes in total gray matter, the correlation of test statistics (an analysis that does not impose dichotomous statistical significance to each gene) showed a significant, moderately positive relationship across diagnoses, suggesting a shared disturbance in mitochondrial function. However, substantially more DEGs were down-regulated in schizophrenia than in bipolar disorder. These differences may reflect lower statistical power in the bipolar disorder group, but it is important to note that the weighted gene coexpression network analysis showed diagnostic differences similar to those in the DEG analyses. In addition, other differences by diagnosis included up-regulated DEGs solely in schizophrenia subjects and the presence of some DEGs in bipolar disorder subjects that were not altered in the schizophrenia group. Together, these data demonstrate that both the severity and the nature of mitochondrial-related gene expression alterations in the DLPFC differ between schizophrenia and bipolar disorder.
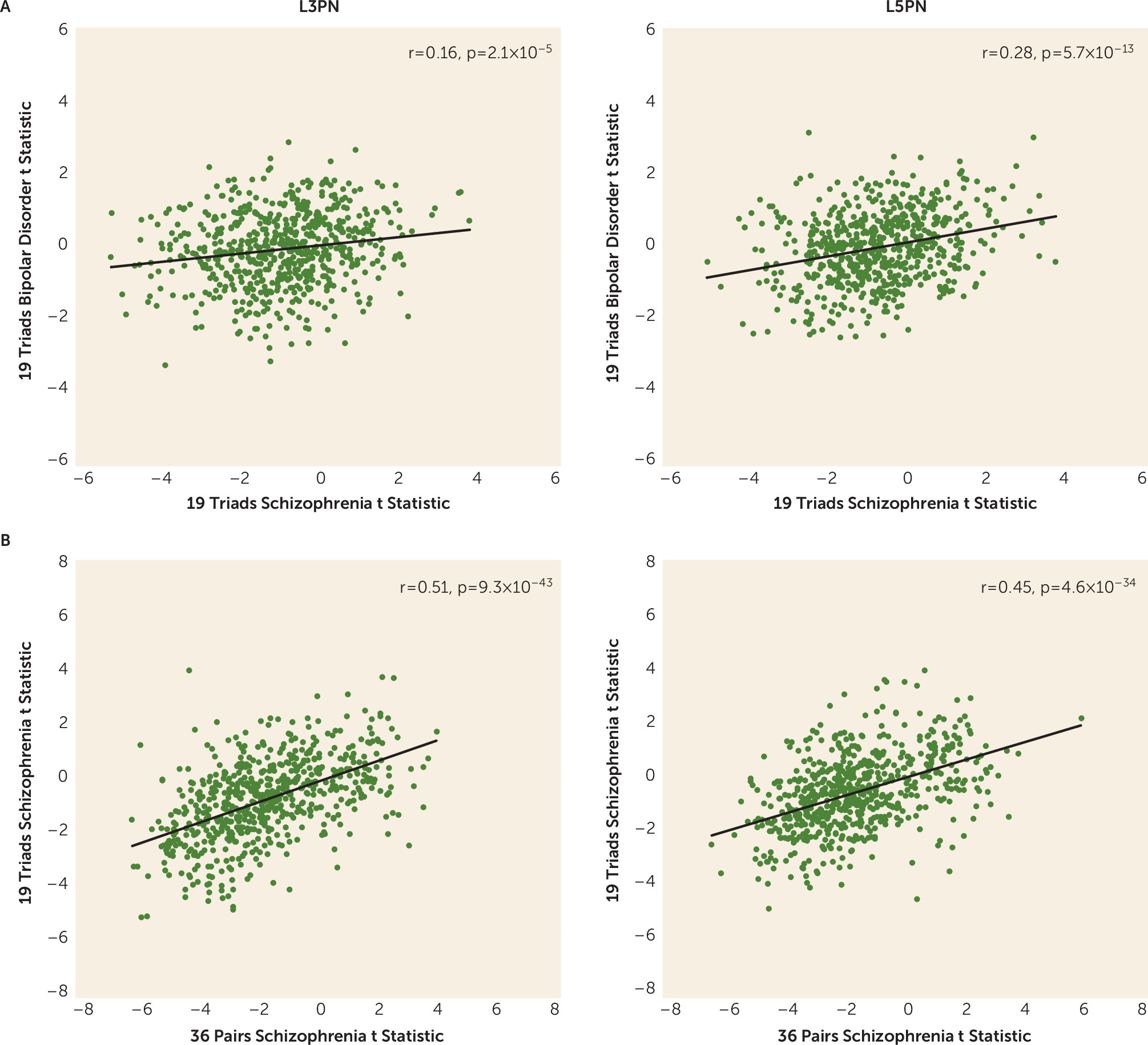
The presence of diagnosis-specific differences in mitochondrial-related gene expression was further supported by the absence of DEGs in L3PNs or L5PNs in bipolar disorder subjects. Because equal numbers of schizophrenia and bipolar disorder subjects were used in these analyses, the diagnostic differences were not confounded by differences in statistical power. Thus, the few DEGs identified in the gray matter of bipolar disorder subjects may reflect alterations in other cell types (
34). For example, the RNA sequencing data reflect gene expression in all cell types, including highly metabolically active cells, such as parvalbumin interneurons. Parvalbumin interneurons have been reported to show down-regulated OXPHOS transcripts in schizophrenia (
19), but studies of this cell type have not been conducted in bipolar disorder subjects. Moreover, the weakly correlated test statistics in each cell population between diagnoses suggests that few of the effects present in schizophrenia are evident in bipolar disorder. In schizophrenia subjects, test statistics were correlated between L3PNs and L5PNs, suggesting that the overall effects of schizophrenia on mitochondrial-related gene expression may be shared across neuronal types in the DLPFC. However, given that multiple up-regulated DEGs were present in gray matter but not in L3PNs or L5PNs in schizophrenia subjects, other neuronal or nonneuronal cell types in the DLPFC may be uniquely affected. Similarly, both neuronal populations also exhibited a unique set of DEGs, which may reflect cell type-specific alterations in mitochondrial-related gene expression resulting from intrinsic differences in the transcriptomes of L3PNs and L5PNs.
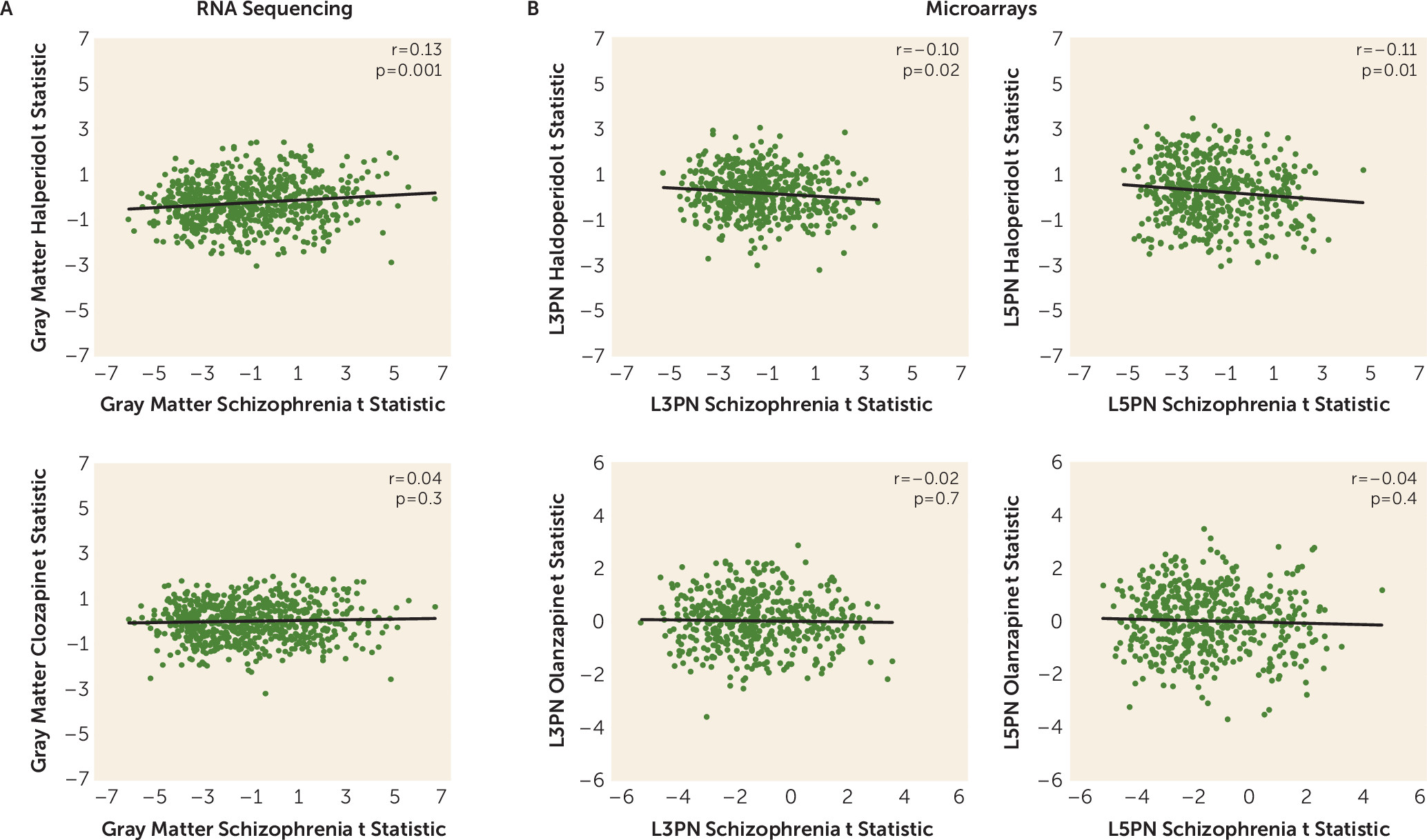
Peripheral metabolic effects of antipsychotic drug treatment, including impaired glucose metabolism, are well documented (reviewed in reference
35), and some cortical neural circuits and mitochondrial characteristics can be affected by chronic treatment with antipsychotic drugs (
25,
26). However, whether mitochondrial-related gene expression, broadly or within specific metabolic pathways, is affected by chronic administration of first- or second-generation antipsychotics was unknown. Our findings that none of the mitochondrial-related genes were detected as differentially expressed at either level of anatomical resolution in monkeys chronically exposed to either first- or second-generation antipsychotics suggest that any effects antipsychotic drugs may have on the mitochondrial-related transcriptome do not occur in DLPFC gray matter or pyramidal neurons. Additionally, mitochondrial-related gene expression and lifetime chlorpromazine-equivalent dose (available for a subset of schizophrenia subjects [
19]) were not significantly correlated in total gray matter, L3PNs, or L5PNs, with the exception of a single gene in L3PNs (RARS2, r=0.72, q=0.01). Thus, any role that antipsychotics may have in altering DLPFC functioning is unlikely to be mediated via changes in mitochondria.
In DLPFC gray matter and L3PNs and L5PNs in schizophrenia subjects, DEGs were significantly enriched for OXPHOS, mitochondrial dysfunction, and sirtuin-signaling pathways. Weighted gene coexpression network analysis showed that the coexpression network modules enriched for these functional pathways were unperturbed in schizophrenia, despite the number of DEGs. This selective and coordinated down-regulated expression of genes related to energy production suggests that schizophrenia is associated with less ATP synthesis via OXPHOS. One mechanism that can result in less OXPHOS is reduced cellular demand for ATP. Indeed, ATP is synthesized in mitochondria only on demand (
36), and neurons normally make coordinated adjustments in the expression of OXPHOS-related genes to meet changes in energetic demand (
37–
40). In neurons, the most energetically demanding processes involve action potential generation and synaptic signaling (
10,
41), and chronically lower rates of neuronal firing cause a coordinated reduction in OXPHOS-related transcripts (
40). These coordinated alterations in OXPHOS-related transcript expression are unlike those identified in response to insults that impair or disrupt the energetic capacity of mitochondria to meet cellular demand (
37,
38). For example, disrupting the expression of electron transport chain complex core subunits, accessory subunits, or assembly factors does not result in a coordinated reduction in the expression of OXPHOS-related transcripts (
42–
47). Moreover, this idea that less neuronal demand for energy production, and not defective mitochondrial function, is operative in the DLPFC in schizophrenia is consistent with existing anatomical and genetic data. For example, L3PNs have a smaller dendritic arbor and lower density of dendritic spines, the primary site of excitatory inputs (
48). Fewer spines suggests that L3PNs receive fewer excitatory synapses in schizophrenia, and their resulting hypoactivity leads secondarily to less excitation of other pyramidal neurons. Indeed, given that L3PNs innervate L5PNs in the DLPFC (
49), less firing of L3PNs is predicted to result in less excitation of this downstream cell population. Additionally, data from genome-wide association studies strongly implicate impaired synaptic processes in the etiology of schizophrenia and not primary insults to mitochondria (
50–
52).
In summary, our analysis across multiple data sets and cohorts of subjects of a large and specific gene set reflecting the multitude of constituent mitochondrial functions in the DLPFC showed a pronounced effect on energy production pathways selectively in schizophrenia subjects. The paucity of findings in bipolar disorder compared with schizophrenia may reflect fundamental differences in the underlying disease processes. Indeed, DLPFC dysfunction and cognitive impairments are thought to be much more prominent in schizophrenia than in bipolar disorder (
53); however, examination of mitochondrial pathways in other brain regions or cell types may identify bipolar disorder-specific alterations. The lower measures of mitochondrial ATP production in schizophrenia may be a consequence of upstream pathological processes that result in less firing of L3PNs and L5PNs, which likely reflects an interaction of genetic vulnerabilities and environmental factors that affect synaptic integrity (
48,
54). As such, therapeutics targeting enhancement of synaptic excitation within this circuit may prove to be beneficial in ameliorating cognitive dysfunction.