Major depressive disorder (MDD) is a leading cause of disability in the United States and worldwide (
1–
3). A limitation of standard-of-care antidepressant therapies for MDD (e.g., selective serotonin reuptake inhibitors) is that they typically require weeks or months to improve depressive symptoms (
4,
5). Furthermore, they are associated with several adverse effects, including weight gain, sexual dysfunction, and cognitive impairment, which may result in treatment discontinuation (
6–
9).
As demonstrated in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, delay in treatment response may be a barrier for some antidepressant therapies and may fuel an extended duration of disability and delay in functional recovery (
4). These therapies may also require daily use for months to years to prevent relapse, potentially resulting in a long-term burden of treatment (
4,
5). Accordingly, some patients with MDD need new treatment options that have a rapid onset of action, high response and remission rates, and minimal safety concerns commonly associated with standard-of-care antidepressant therapies.
To address this unmet need, therapies with novel mechanisms of action are being investigated, including therapies targeting γ-aminobutyric acid (GABA) signaling. GABA is the primary inhibitory neurotransmitter in the brain and is thought to serve an important role in network signaling by balancing excitatory and inhibitory neurotransmission (
10–
12). Patients with depression may have impaired network balance and low or reduced plasma and cerebrospinal fluid GABA levels (
13–
16). However, the therapeutic potential of modulating GABA signaling for the treatment of MDD is underexplored.
Inhibitory GABA receptor signaling is modulated by endogenous neuroactive steroids such as allopregnanolone, which acts as a positive allosteric modulator of both synaptic and extrasynaptic GABA type A receptors (GABA
ARs) (
17). Supporting the potential of modulating GABA
AR signaling for the treatment of depression, brexanolone, a proprietary intravenous formulation of allopregnanolone, is currently the only drug approved by the U.S. Food and Drug Administration (FDA) specifically for the treatment of postpartum depression in patients age 15 or older (
18–
21). Phase 3 studies of brexanolone demonstrated a rapid onset of effect in patients with postpartum depression, with significant placebo-adjusted improvements in depressive symptoms observed at 60 hours following a single intravenous infusion of brexanolone (
19). Zuranolone, a positive allosteric modulator of synaptic and extrasynaptic GABA
ARs, is thought to upregulate GABA
AR expression and enhance inhibitory GABAergic signaling. As a neuroactive steroid, zuranolone is hypothesized to rapidly restore network balance in brain areas dysregulated in depression (
17,
22,
23). In a preclinical study using the beta EEG band (β band) as a robust biomarker of GABA
AR positive allosteric modulator activity, oral administration of zuranolone in mice led to a rapid (<1 hour) increase in β band power, suggesting a potential rapid onset of activity in the brain (
23).
The LANDSCAPE and NEST clinical development programs include a series of randomized clinical trials assessing the efficacy and safety of zuranolone as an oral, once-daily, 14-day therapy for MDD and postpartum depression, respectively. Previous studies in adult patients with MDD (phase 2) and postpartum depression (phase 3) met their primary endpoint of a statistically significant improvement in depressive symptoms with zuranolone 30 mg/day compared with placebo at day 15, as assessed by total score on the 17-item Hamilton Depression Rating Scale (HAM-D) (
24,
25). Here, we report the results of a phase 3 study (ClinicalTrials.gov identifier: NCT04442490) evaluating the efficacy, safety, and tolerability of zuranolone 50 mg/day for the treatment of MDD in adults.
Methods
Study Design and Participants
This was a multicenter, randomized, double-blind, placebo-controlled phase 3 clinical trial conducted between May 2020 and April 2021 at 39 sites in the United States. All patients were screened ≤28 days prior to day 1 of the study. The study received institutional review board approval at each site, was conducted in accordance with the ethical guidelines of the Declaration of Helsinki, and was consistent with the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use and Good Clinical Practice. The 50-mg/day dosage used in the study was selected based on direct and modeled data to assess efficacy and safety outcomes of zuranolone at higher dosages (further details are provided in the Supplemental Methods section of the online supplement). Patients were 18–64 years old with a diagnosis of MDD (per Structured Clinical Interview for DSM-5 criteria) with symptoms for ≥4 weeks and a HAM-D score ≥24 at screening and prior to dosing on day 1. The study excluded patients with active psychosis, those who attempted suicide or were at risk of suicide in their current MDD episode, and those with treatment-resistant depression, defined in this study as persistent depressive symptoms despite treatment within the current major depressive episode with antidepressants (excluding antipsychotics) from two different classes at adequate dosages for ≥4 weeks. A full list of eligibility criteria is provided in the Supplemental Methods section of the online supplement. After screening, eligible patients were randomized to treatment with zuranolone 50 mg/day or placebo and underwent a 14-day treatment period and 28-day follow-up period (see Figure S1 in the online supplement). The use of antidepressants in both groups was permitted, provided that patients were on a stable dosage for ≥60 days prior to day 1 and agreed to continue on the stable dosage through day 42. All patients provided written informed consent after receiving a complete description of the study.
Randomization and Blinding
Randomization, in a 1:1 ratio, was performed centrally via an interactive response technology system. Patients, clinicians, site personnel, the study sponsor, and the study team were blinded to treatment allocation. Blinding was maintained until database lock after all patients completed the study visit at day 42.
Procedures
Patients self-administered oral zuranolone 50 mg or placebo once daily in the evening with fat-containing food. Study visits occurred at days 1, 3, 8, 12, 15, 18, 21, 28, 35, and 42. All reported assessments were administered by the investigators. The Columbia–Suicide Severity Rating Scale (C-SSRS) was used to evaluate suicidal ideation or behavior. HAM-D score, Clinical Global Impressions severity (CGI-S) score, Montgomery-Åsberg Depression Rating Scale (MADRS) score, and CGI improvement (CGI-I) score were used to evaluate depressive symptoms. The Hamilton Anxiety Rating Scale (HAM-A) score was used to evaluate symptoms of anxiety, and the 20-item Physician’s Withdrawal Checklist (PWC-20) score was used to assess symptoms of withdrawal. Further details on study procedures and visits are provided in the Supplemental Methods section of the online supplement.
Outcome Measures
The primary endpoint was change from baseline in HAM-D score at day 15. Key secondary endpoints were change from baseline in CGI-S score at day 15 and change from baseline in HAM-D score at days 3, 8, and 42. Other secondary endpoints included the proportion of patients who achieved HAM-D response (a reduction ≥50% in HAM-D score from baseline) and remission (a HAM-D score ≤7) at days 15 and 42, a CGI-I response of “much improved” or “very much improved” at day 15, change from baseline in MADRS score at day 15, and change from baseline in HAM-A score at day 15. Subgroup analyses included change from baseline in HAM-D score by baseline patient characteristics and HAM-D response and remission rates by antidepressant use at baseline.
Safety was evaluated from the incidence and severity of treatment-emergent adverse events and serious adverse events. Additional safety assessments included suicidal ideation and behavior (per the C-SSRS), potential withdrawal symptoms after zuranolone discontinuation (per the PWC-20), and changes in laboratory values, ECG, and vital signs.
Statistical Analysis
Based on a two-sided alpha level of 0.05 and an assumed 10% dropout rate, a sample size of 240 patients was required to have 90% power if the true placebo-adjusted treatment difference in the primary endpoint of HAM-D score was 4 points (assuming a standard deviation of 9 points). To increase power for analyses of key secondary endpoints, the sample size was adjusted to permit enrollment of approximately 575 patients (randomized set). The safety set included all patients who received at least one dose of zuranolone or placebo. The full analysis set, used for efficacy analyses, included all patients in the safety set who had a valid baseline HAM-D score and at least one postbaseline HAM-D score.
Primary and secondary change from baseline endpoints were analyzed in the full analysis set with mixed models for repeated measures (MMRM) comparing the change from baseline least squares mean (LSM) differences in outcome measures between the zuranolone and placebo groups. For each outcome measure (e.g., change from baseline in HAM-D score and in CGI-S score), one MMRM model was used to estimate all time points. The models included terms for treatment, time point, baseline outcome measure, antidepressant use at baseline (yes or no), and the treatment-by-time point interaction. All explanatory variables were treated as fixed effects, and random effects were included as part of the marginal covariance matrix (
26). Each model used an unstructured covariance matrix to model the within-patient errors. A generalized estimating equation (GEE) method was used to analyze HAM-D response, HAM-D remission, and CGI-I response. An unstructured covariance matrix was used for each GEE model to account for the within-patient error. Multiplicity adjustment for statistical hypothesis testing of the key secondary endpoints was conducted using a prespecified fixed sequence reviewed by the FDA prior to database lock and final analysis. Key secondary endpoints were tested only if the primary endpoint demonstrated a statistically significant difference at the 0.05 level. The sequence of key secondary endpoint testing began with the change from baseline in CGI-S score at day 15, followed by the change from baseline in HAM-D score at days 8, 3, and 42, in that order. Formal testing stopped if the p value for the previous endpoint in the sequence was >0.05, and subsequent endpoints were interpreted only with nominal p values. All other secondary endpoints and additional analyses were not adjusted for multiplicity and hence were interpreted with a nominal p value. The effect size was measured using Cohen’s d estimates for change from baseline in HAM-D score at days 3, 8, 12, and 15.
Results
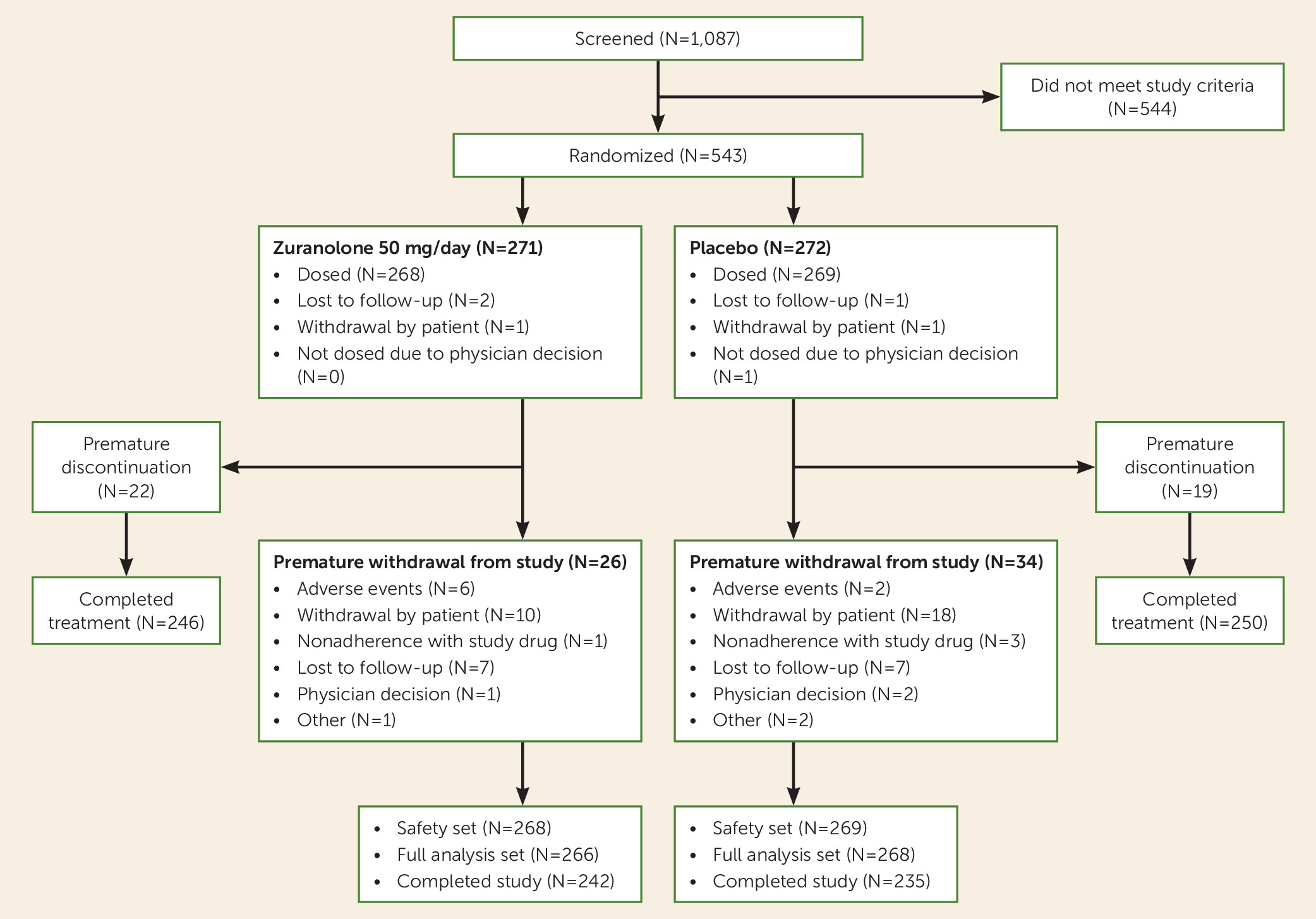
Of 1,087 patients screened, 543 were randomized, 537 were included in the safety set (268 in the zuranolone group, 269 in the placebo group), and 534 were included in the full analysis set (266 in the zuranolone group, 268 in the placebo group) (
Figure 1). Baseline characteristics were generally balanced between the zuranolone and placebo groups (
Table 1). Patients in both groups had a diagnosis of MDD for a mean of approximately 11 years, with a mean duration of >1 year for the current depressive episode, and approximately 30% were on antidepressant therapies (
Table 1; see Table S1 in the
online supplement for concomitant antidepressant therapies). Most patients were White (zuranolone group, 63.1%; placebo group, 76.6%) and female (zuranolone group, 69.4%; placebo group, 61.7%). Patients had similar severity of depressive symptoms as assessed by the HAM-D (mean scores: zuranolone group, 26.8; placebo group, 26.9).
Efficacy
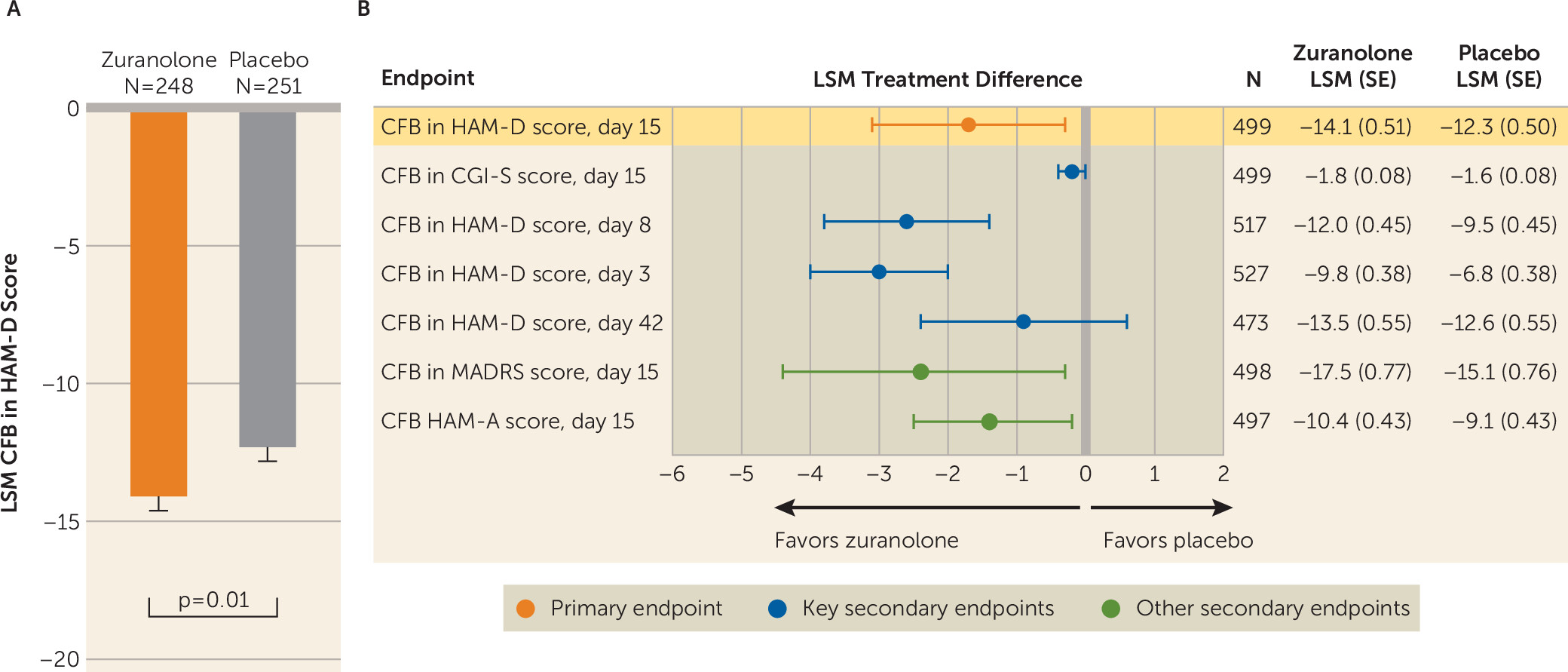
Treatment with zuranolone demonstrated a significantly greater improvement in depressive symptoms compared with placebo at day 15, as assessed by change from baseline in HAM-D score (LSM change, −14.1 [SE=0.5] vs. −12.3 [SE=0.5], p=0.01; Cohen’s d=0.23) (
Figure 2,
Figure 3A; see effect size data in Table S2 in the
online supplement). The LSM change from baseline in CGI-S score at day 15 was numerically greater for zuranolone compared with placebo, but the difference was not statistically significant (−1.8 [SE=0.1] vs. −1.6 [SE=0.1], p=0.12), so formal testing of the following key secondary endpoints in the fixed sequence stopped, and all subsequent key secondary endpoints were interpreted with nominal p values (
Figure 2B; see also Figure S2 in the
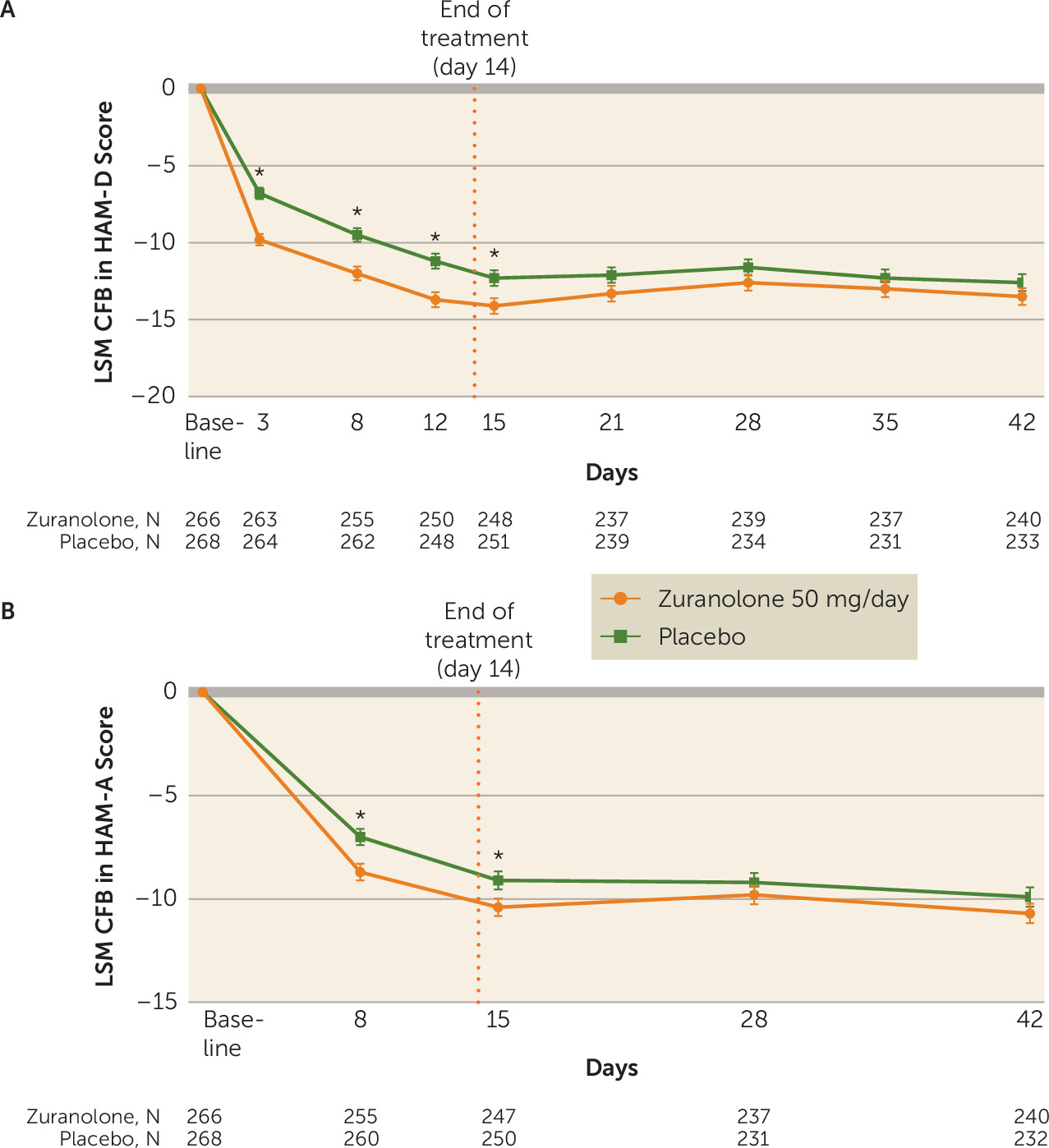
online supplement). In addition to improvements in depressive symptoms demonstrated at day 15, the LSM change from baseline in HAM-D score was numerically greater for zuranolone compared with placebo at all other study visits, including day 3 (−9.8 [SE=0.4] vs. −6.8 [SE=0.4], p<0.001; Cohen’s d=0.49), day 8 (−12.0 [SE=0.5] vs. −9.5 [SE=0.5], p<0.001; Cohen’s d=0.38), day 12 (−13.7 [SE=0.5] vs. −11.2 [SE=0.5], p<0.001; Cohen’s d=0.32), day 21 (−13.3 [SE=0.5] vs. −12.1 [SE=0.5], p=0.09), day 28 (−12.6 [SE=0.5] vs. −11.6 [SE=0.5], p=0.18), day 35 (−13.0 [SE=0.6] vs. −12.3 [SE=0.6], p=0.37), and day 42 (−13.5 [SE=0.6] vs. −12.6 [SE=0.6], p=0.23) (
Figure 3A; see effect size data in Table S2 in the
online supplement).
The subgroup analyses (based on baseline characteristics) were generally consistent with the results of the primary endpoint, showing favorable results for zuranolone compared with placebo (see Figure S3 in the
online supplement). As seen with the primary endpoint, greater improvement in depressive symptoms was observed for zuranolone at day 15 compared with placebo as measured by change from baseline in MADRS score (LSM change, −17.5 [SE=0.8] vs. −15.1 [SE=0.8], p=0.02) (
Figure 2B), which was maintained at all posttreatment visits (see Figure S4 in the
online supplement). Greater improvements in anxiety symptoms at day 15 were observed for zuranolone compared with placebo, as assessed by change from baseline in HAM-A score (LSM change, −10.4 [SE=0.4] vs. −9.1 [SE=0.4], p=0.02) (
Figure 2B). Numerical but nonsignificant improvements in HAM-A score were maintained with zuranolone compared with placebo at day 28 (LSM change, −9.8 [SE=0.5] vs. −9.2 [SE=0.5], p=0.31) and day 42 (LSM change, −10.7 [SE=0.5] vs. −9.9 [SE=0.5], p=0.21) (
Figure 3B). A greater proportion of patients receiving zuranolone achieved CGI-I response (ratings of “much improved” or “very much improved”) compared with patients receiving placebo at day 3 (30.0% [79/263] vs. 18.9% [50/264]; odds ratio=1.9, 95% CI=1.2–2.8; p=0.003), at day 15, and at all study visits throughout the posttreatment follow-up period (see Figure S5 in the
online supplement).
A greater proportion of patients receiving zuranolone than patients receiving placebo achieved HAM-D response at day 3 (29.3% [77/263] vs. 16.3% [43/264]; odds ratio=2.1, 95% CI=1.4–3.3; p<0.001), at day 15 (56.0% [139/248] vs. 47.0% [118/251]; odds ratio=1.4, 95% CI=1.0–2.0; p=0.06), and at day 42 (52.9% [127/240] vs. 45.9% [107/233]; odds ratio=1.4, 95% CI=1.0–1.9; p=0.09) (see Figure S6A in the online supplement). A small proportion of patients receiving either zuranolone or placebo who were assessed for HAM-D response at both days 15 and 42 had a HAM-D response at day 15 but not at day 42 (15.2% and 12.9%, respectively). Rates of HAM-D remission were observed in a greater proportion of patients receiving zuranolone at day 3 compared with patients who received placebo (7.6% [20/263] vs. 2.3% [6/264]; odds ratio=3.6, 95% CI=1.4–9.1; p=0.01) (see Figure S7A in the online supplement). Remission rates were numerically but not significantly greater in the zuranolone group at day 15 (29.8% [74/248] vs. 27.1% [68/251]; odds ratio=1.1, 95% CI=0.8–1.7; p=0.55) and at day 42 (30.8% [74/240] vs. 29.6% [69/233]; odds ratio=1.1, 95% CI=0.7–1.6; p=0.68) (see Figure S7A in the online supplement). Greater HAM-D response and remission rates were observed among patients receiving zuranolone compared with placebo irrespective of concomitant antidepressant use from baseline (see Figures S6B,C and S7B,C in the online supplement).
Safety
The overall incidence of treatment-emergent adverse events with zuranolone was 60.1% (161/268), compared with 44.6% (120/269) with placebo (
Table 2). Most patients who experienced treatment-emergent adverse events had events that were at most mild or moderate (zuranolone, 95.0% [153/161]; placebo, 97.5% [117/120]). The most common treatment-emergent adverse events (>5% frequency in any group) were somnolence, dizziness, headache, sedation, and diarrhea (
Table 2). A total of 23 patients in the zuranolone group and one in the placebo group had their dosage reduced due to treatment-emergent adverse events (
Table 2), the most common being dizziness and somnolence with zuranolone and fatigue with placebo. Nine patients in the zuranolone group and four in the placebo group discontinued treatment due to treatment-emergent adverse events (
Table 2). The most common treatment-emergent adverse events that led to treatment discontinuation were nervous system disorders, including dizziness and sedation in the zuranolone group and sedation and somnolence in the placebo group. The incidence of treatment-emergent adverse events was not notably different among patients receiving zuranolone with or without a concomitant antidepressant therapy at baseline (see Table S3 in the
online supplement). No deaths, loss of consciousness, weight gain, sexual dysfunction, or euphoria were reported in either treatment arm. Four patients (0.7%), two in each group, experienced serious adverse events. Two patients, one in each group, experienced serious adverse events during the treatment period; the remaining serious adverse events occurred during the posttreatment follow-up period. Details of serious adverse events are provided in the Supplemental Results section of the
online supplement.
There were no changes in laboratory values, ECG results, or vital signs leading to treatment interruption or discontinuation in either group. No evidence of increased suicidal ideation or behavior relative to the baseline assessment was observed in either the zuranolone or the placebo group, as measured by the C-SSRS (see Table S4 in the online supplement). There was no indication of increased withdrawal symptoms after zuranolone treatment (see the online supplement).
Discussion
In this study, patients receiving zuranolone 50 mg/day demonstrated significantly greater improvements in depressive symptoms at day 15 compared with those receiving placebo, and the observed onset of effect was rapid, with greater improvement in HAM-D and CGI-S scores observed for zuranolone compared with placebo at the earliest assessment, on day 3. The effect sizes for change from baseline in HAM-D score at visits from day 3 to day 15 ranged from 0.49 to 0.23, which are comparable to effect sizes seen at the primary assessment time point in clinical studies of approved antidepressant therapies (a mean effect size of 0.29 from 34 registration studies conducted after 2000) (
27). The differences in HAM-D score between the zuranolone and placebo groups at study visits from day 3 (−3.0) through day 15 (−1.7) also surpassed the estimated minimal important difference (−1.67), suggesting that the improvements in depressive symptoms were clinically meaningful (
28). Greater improvements in depressive and anxiety symptoms with zuranolone were sustained in the follow-up period, as evidenced by the generally stable HAM-D, MADRS, and HAM-A scores across all study visits through day 42. Furthermore, while the sample sizes were relatively small, the efficacy results favored zuranolone regardless of the use of antidepressant therapies at baseline. The rapid onset of action and potential for benefit of zuranolone with an antidepressant therapy is further reinforced by a recently completed phase 3 study of patients with MDD (ClinicalTrials.gov identifier: NCT04476030), which showed improvements in patients co-initiated with zuranolone and a standard-of-care antidepressant therapy compared with patients co-initiated with placebo and a standard-of-care antidepressant therapy at day 3 and over the treatment period through day 15.
Consistent with previous studies of zuranolone at 30 mg/day, no weight gain, sexual dysfunction, withdrawal symptoms, or increased suicidal ideation or behavior was observed in this study. Additionally, no new safety findings emerged with zuranolone 50 mg/day compared with previous studies of zuranolone 30 mg/day (
24,
25). Zuranolone 50 mg/day was generally well tolerated, with most treatment-emergent adverse events being mild or moderate in severity and low rates of dosage reduction or study discontinuation due to treatment-emergent adverse events. In the present study and the published studies of zuranolone to date, the most common adverse events included somnolence and headache and were generally not clinically serious (
24,
25). As efficacy and safety outcomes in this study were not notably different between patients receiving zuranolone with or without an antidepressant therapy, these data support zuranolone as a viable option as a monotherapy or concomitant with an antidepressant therapy in patients with MDD, if approved.
The approach of targeting GABA
ARs, although emergent for the treatment of depression, is well characterized for the treatment of anxiety-related disorders. Benzodiazepines are commonly used to treat anxiety and act on synaptic GABA
ARs to promote phasic inhibition. By contrast, zuranolone has a different binding site and acts on both synaptic and extrasynaptic GABA
ARs, leading to potentiation of both phasic and tonic inhibition, respectively (
10,
11,
22,
23,
29–
31). This mechanistic difference of promoting both transient phasic and sustained tonic inhibition may help restore normal network function in the brain (
24,
25). The difference between the hypothesized mechanism of action of neuroactive steroids, such as zuranolone, and that of benzodiazepines may also explain the observation that while benzodiazepines promote receptor desensitization, downregulation, and tolerance, neuroactive steroids in preclinical studies upregulated receptor expression (
32,
33).
Patients with MDD frequently have symptoms of anxiety stemming from their depression and may receive treatment with a standard-of-care antidepressant therapy plus a benzodiazepine (
34,
35). This is because benzodiazepines elicit a rapid onset of effect and may work with an antidepressant therapy to quickly alleviate symptoms of anxiety and depression and prevent discontinuation of antidepressant therapy (
36). However, the results supporting this strategy are mixed, and evidence suggests that the potential additive effects may not persist with long-term treatment (
37). Furthermore, benzodiazepine monotherapy is associated with limited antidepressant effects, and the potential benefits of the antidepressant-benzodiazepine combination may be offset by the risk of tolerance, dependence, and abuse associated with benzodiazepines, and an increased likelihood of experiencing adverse events (
37–
40). The improvements observed in both HAM-D and HAM-A scores in the present study suggest that patients with MDD with elevated anxiety who received zuranolone experienced improvements in anxiety symptoms compared with placebo. Long-term outcomes of using zuranolone monotherapy for management of MDD with elevated anxiety are not yet known and will be further elucidated with real-world use of zuranolone, if approved.
A limitation of this study is that it was a short-term one designed to assess the outcomes of patients after one 14-day treatment course of zuranolone. The long-term safety and efficacy of zuranolone and the potential need for repeated treatment courses over a 1-year period are being addressed in an ongoing open-label study (ClinicalTrials.gov identifier: NCT03864614). The present study’s patient population was also severely depressed at study entry (mean baseline HAM-D scores of 26.8 and 26.9 for patients receiving zuranolone and placebo, respectively), which may limit the degree to which the results generalize to patients with mild or moderate depressive symptoms. However, the mean baseline HAM-D score in this study is consistent with a meta-analysis of several clinical trials of standard-of-care antidepressant therapies, which reported mean baseline HAM-D scores ranging from 17.0 to 30.5, and other studies in the LANDSCAPE program included patients with moderate to severe depressive symptoms (
41). While the patient population was racially and ethnically diverse in both groups, there was a slightly lower proportion of White patients and a higher proportion of African American and Asian patients in the zuranolone group compared with the placebo group, which may limit the generalizability of the results. Additionally, this study showed a robust placebo response, and its magnitude was similar to that observed previously for zuranolone or antidepressant therapies. The observed placebo response may be due in part to the high frequency of study visits (10 visits in 42 days), which is both higher than the typical frequency of real-world visits and consistent with previous evaluations in other standard-of-care antidepressant therapy trials, which showed a positive correlation between the number of study visits and placebo response (
42,
43). Another factor that possibly had an influence on the results was that the study was conducted entirely during the COVID-19 pandemic. A U.S. population-based survey conducted at the beginning of the COVID-19 pandemic demonstrated a threefold increase in depressive symptoms as measured by the Patient Health Questionnaire–9 compared with prepandemic levels (
44). The frequent in-person visits in this study may have contributed to alleviating feelings of isolation and predisposed patients to experience improvements in symptoms even if they were receiving placebo.
An important observation in this study was that a small proportion of patients who had a HAM-D response at day 15 did not maintain the HAM-D response at day 42, after cessation of treatment. Accordingly, clinicians may continue to monitor patients after response to the initial treatment to determine whether the major depressive episode was successfully treated and continue to intervene if appropriate.