Pathogenic Variants and Ascertainment: Neuropsychiatric Disease Risk in a Health System Cohort
Publication: American Journal of Psychiatry
Our understanding of genetic risk for neurodevelopmental disorders is based on studies of individuals clinically ascertained for specific diagnoses. While large-scale studies have identified hundreds of genes and rare variants for phenotypes such as autism, intellectual disability, and schizophrenia, a large fraction of the identified genetic variants are nonspecific and variable in both prevalence and degree of affectedness. Akin to the parable of the four blind men describing an elephant, studies on independently ascertained disease cohorts have often implicated the same genetic cause for distinct disorders. For example, mutations disrupting SCN2A have been independently reported in cohorts manifesting epilepsy, schizophrenia, and autism (1), and deletions on chromosome 15q13.3 have been associated with epilepsy, schizophrenia, and intellectual disability while further being documented in apparently unaffected individuals (2). In fact, many disease-associated copy number variants—that is, deletions and duplications of genomic regions—have also been reported in unselected populations, where carriers of those variants were found to manifest subclinical psychiatric and cognitive defects (3, 4). The variable expressivity of genomic variants that were originally believed to be highly penetrant toward a specific neurodevelopmental phenotype reveals that our current notion of disease pathogenicity is confounded by ascertainment bias of studying affected individuals from clinical cohorts.
In this issue of the Journal, Shimelis and colleagues (5) assess the impact of pathogenic mutations on neuropsychiatric outcomes in over 90,000 individuals from the Geisinger health care system population (termed the DiscovEHR cohort). Focusing on a curated list of 94 genes implicated in neurodevelopmental and psychiatric features, the authors analyzed exome sequencing data and found that 312 (0.34%) individuals carried a rare loss-of-function variant. The observed prevalence of pathogenic mutations was several times lower than prevalences reported in disease cohorts (6). Although more representative of the general population than a disease-specific cohort, cohorts derived from health care systems may overrepresent individuals with a clinical diagnosis and hence show a higher prevalence of pathogenic variants than an unselected general population (Figure 1). In fact, the prevalence of pathogenic copy number variants in the DiscovEHR cohort was higher compared to that of individuals from the general Icelandic population (4), healthy aging adults in the UK Biobank (7), or participants in the Estonian Biobank studies (8). While survivor bias, a type of selection bias arising from healthy volunteers participating in cohorts such as the UK Biobank, could account for some of these differences, prevalence estimates in unselected populations could inform us on the selective effects of rare variants and their disease relevance in a clinical context.
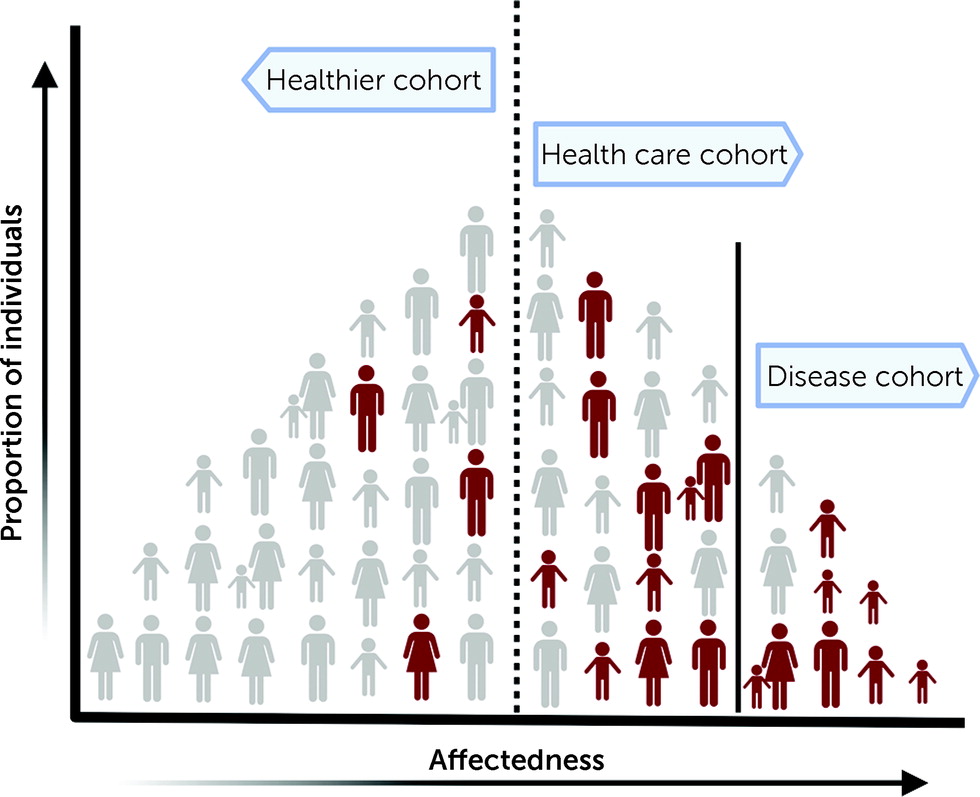
FIGURE 1. Schematic representation of prevalence of pathogenic variants in different cohortsa
aThe individuals in red carry rare pathogenic variants associated with neurodevelopmental and psychiatric disorders. The Shimelis et al. study and previous studies on different populations show that prevalence and penetrance rates of pathogenic variants vary across different ascertainments.
To assess the penetrance of the pathogenic variants, the authors identified individuals with variants who also had an ICD-10 diagnosis for 14 neurodevelopmental and psychiatric features. Only 34% of individuals with a pathogenic variant had an ICD-10 diagnosis. This is noteworthy, as the 94 genes selected in this study are mostly known to be associated with highly penetrant disorders, typically diagnosed in early childhood. The authors identified pathogenic mutations in 61 of the 94 genes, including those associated with a range of neuropsychiatric outcomes, such as ANK2 (9) and SHANK2 (10). Some genes recapitulated known associations, such as SCN1A and STXBP1 for epilepsy (11, 12), while other genes showed novel associations with previously unreported phenotypes. For example, epilepsy in an individual with a mutation in RAI1, the causative gene for Smith-Magenis syndrome (13), and depressive disorder in individuals with mutations in KMT2D or NSD1, genes associated with Kabuki syndrome (14) and Sotos syndrome (15), respectively, were noted. Some pathogenic variants may also show pleiotropic effects, with individuals manifesting features outside cognitive and behavioral domains, such as cardiac or renal defects, due to shared genetic etiologies (16, 17). Indeed, Shimelis et al. found that about 11% of individuals with pathogenic variants had congenital anomalies, with 5.4% manifesting cardiac or renal disorders. When the authors included the diagnosis of anxiety and depression or a history of congenital malformation in their analysis, their overall penetrance estimates increased to 68.6% and 71.2%, respectively. These observations indicate that genes that typically cause syndromic diagnoses in affected children can be associated with milder neuropsychiatric or unexpected pleiotropic features in a broadly ascertained adult population.
Although ICD-based diagnosis may not capture the entire spectrum of phenotypic effects, the substantially reduced penetrance of pathogenic variants detected in this study has significant implications for understanding the complexity of genetic disorders. The role of the genetic background, which is a collection of variants of different classes and frequencies that co-occur with the primary pathogenic variant, in modulating disease risk across ascertainments cannot be overstated. The effects of naturally occurring genetic variants on behavioral traits have been well documented in tractable model systems, such as mice and flies (18). For example, Sittig et al. (19) found significant variation in behavioral phenotypes when mice lacking bipolar-associated genes were tested under different strain-specific genetic backgrounds. A multi-hit model has been proposed and explored in the context of variably expressive variants in humans, in which a primary variant sensitizes the genome toward risk for a range of neuropsychiatric outcomes and its interplay with other variants determines the trajectories toward distinct disorders (20). For example, Davies et al. (21) found a substantial contribution of schizophrenia polygenic risk scores toward both cognitive defects and schizophrenia among individuals with 22q11.2 deletion. Another implication of low-penetrance variants is their high likelihood of being transmitted across generations. These variants may cause mild features, as observed in this study, but in conjunction with other rare variants may cause more severe disease in subsequent generations. For example, a recent study showed that individuals having pathogenic mutations in two or three genes were more likely to manifest intellectual disability than those with mutations in individual genes (22). While these models explain increased disease risk, the reduced penetrance of pathogenic mutations observed by Shimelis et al. could in part be due to a protective effect of other variants in the genetic background that may alleviate risk for disease (23). For example, Backman et al. (24) found that a missense variant in ST6GALNAC5 within the UK Biobank cohort was associated with protection against lower gray–white matter contrast, a feature linked with an increased rate of cognitive decay. Protective alleles are generally harder to find because of their rarity, but population-scale genetic studies show promise in this area and could increase the list of potential therapeutic targets relevant to disease. As illustrated by the Shimelis et al. study, analyzing populations not selected for a specific disorder has enabled reevaluation of disease gene pathogenicity within the context of the genetic background.
Currently, a majority of the efforts to understand disease etiology are concentrated on identifying high-effect-size rare variants and individual or collective effects of common variants, but it is becoming increasingly clear that disease penetrance is a result of an interplay between pathogenic mutations and the genetic background. Population-based biobanks provide an unbiased representation of the entire spectrum of genomic variation and increased power to identify genetic associations for complex disorders. Insights derived from population-based studies of neuropsychiatric disorders will allow for a genotype-first approach to diagnosis, timely intervention due to better understanding of disease prognosis, and identification of drug targets and treatment options owing to an improved perception of disease mechanisms.
References
1.
Reynolds C, King MD, Gorman KM: The phenotypic spectrum of SCN2A-related epilepsy. Eur J Paediatr Neurol 2020; 24:117–122
2.
Girirajan S, Eichler EE: Phenotypic variability and genetic susceptibility to genomic disorders. Hum Mol Genet 2010; 19:R176–R187
3.
Kendall KM, Bracher-Smith M, Fitzpatrick H, et al: Cognitive performance and functional outcomes of carriers of pathogenic copy number variants: analysis of the UK Biobank. Br J Psychiatry 2019; 214:297–304
4.
Stefansson H, Meyer-Lindenberg A, Steinberg S, et al: CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 2014; 505:361–366
5.
Shimelis H, Oetjens MT, Walsh LK, et al: Prevalence and penetrance of rare pathogenic variants in neurodevelopmental psychiatric genes in a health care system population. Am J Psychiatry 2023; 180:65–72
6.
Rolland T, Cliquet F, Anney RJL, et al: Sub-diagnostic effects of genetic variants associated with autism. medRxiv, April 11, 2022. https://www.medrxiv.org/content/10.1101/2021.02.12.21251621v3
7.
Crawford K, Bracher-Smith M, Owen D, et al: Medical consequences of pathogenic CNVs in adults: analysis of the UK Biobank. J Med Genet 2019; 56:131–138
8.
Männik K, Mägi R, Macé A, et al: Copy number variations and cognitive phenotypes in unselected populations. JAMA 2015; 313:2044–2054
9.
Stevens SR, Rasband MN: Ankyrins and neurological disease. Curr Opin Neurobiol 2021; 69:51–57
10.
Leblond CS, Nava C, Polge A, et al: Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet 2014; 10:e1004580
11.
Escayg A, MacDonald BT, Meisler MH, et al: Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 2000; 24:343–345
12.
Saitsu H, Kato M, Mizuguchi T, et al: De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet 2008; 40:782–788
13.
Slager RE, Newton TL, Vlangos CN, et al: Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet 2003; 33:466–468
14.
Ng SB, Bigham AW, Buckingham KJ, et al: Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 2010; 42:790–793
15.
Kurotaki N, Imaizumi K, Harada N, et al: Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet 2002; 30:365–366
16.
Homsy J, Zaidi S, Shen Y, et al: De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015; 350:1262–1266
17.
Sanna-Cherchi S, Westland R, Ghiggeri GM, et al: Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest 2018; 128:4–15
18.
Flint J, Mackay TFC: Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res 2009; 19:723–733
19.
Sittig LJ, Carbonetto P, Engel KA, et al: Genetic background limits generalizability of genotype-phenotype relationships. Neuron 2016; 91:1253–1259
20.
Pizzo L, Jensen M, Polyak A, et al: Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet Med 2019; 21:816–825
21.
Davies RW, Fiksinski AM, Breetvelt EJ, et al: Using common genetic variation to examine phenotypic expression and risk prediction in 22q11.2 deletion syndrome. Nat Med 2020; 26:1912–1918
22.
Pounraja VK, Girirajan S: A general framework for identifying oligogenic combinations of rare variants in complex disorders. Genome Res 2022 32:904–915
23.
Harper AR, Nayee S, Topol EJ: Protective alleles and modifier variants in human health and disease. Nat Rev Genet 2015; 16:689–701
24.
Backman JD, Li AH, Marcketta A, et al: Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 2021; 599:628–634
Information & Authors
Information
Published In


History
Accepted: 10 November 2022
Published online: 1 January 2023
Published in print: January 01, 2023
Keywords
Authors
Competing Interests
The authors report no financial relationships with commercial interests.
Funding Information
Research by Dr. Girirajan is supported by NIH grants GM121907 and NS122398 and by resources from Pennsylvania State University.
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBMedia
Figures
FIGURE 1. Schematic representation of prevalence of pathogenic variants in different cohortsa
aThe individuals in red carry rare pathogenic variants associated with neurodevelopmental and psychiatric disorders. The Shimelis et al. study and previous studies on different populations show that prevalence and penetrance rates of pathogenic variants vary across different ascertainments.
Other
Tables
References
References
1.
Reynolds C, King MD, Gorman KM: The phenotypic spectrum of SCN2A-related epilepsy. Eur J Paediatr Neurol 2020; 24:117–122
2.
Girirajan S, Eichler EE: Phenotypic variability and genetic susceptibility to genomic disorders. Hum Mol Genet 2010; 19:R176–R187
3.
Kendall KM, Bracher-Smith M, Fitzpatrick H, et al: Cognitive performance and functional outcomes of carriers of pathogenic copy number variants: analysis of the UK Biobank. Br J Psychiatry 2019; 214:297–304
4.
Stefansson H, Meyer-Lindenberg A, Steinberg S, et al: CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 2014; 505:361–366
5.
Shimelis H, Oetjens MT, Walsh LK, et al: Prevalence and penetrance of rare pathogenic variants in neurodevelopmental psychiatric genes in a health care system population. Am J Psychiatry 2023; 180:65–72
6.
Rolland T, Cliquet F, Anney RJL, et al: Sub-diagnostic effects of genetic variants associated with autism. medRxiv, April 11, 2022. https://www.medrxiv.org/content/10.1101/2021.02.12.21251621v3
7.
Crawford K, Bracher-Smith M, Owen D, et al: Medical consequences of pathogenic CNVs in adults: analysis of the UK Biobank. J Med Genet 2019; 56:131–138
8.
Männik K, Mägi R, Macé A, et al: Copy number variations and cognitive phenotypes in unselected populations. JAMA 2015; 313:2044–2054
9.
Stevens SR, Rasband MN: Ankyrins and neurological disease. Curr Opin Neurobiol 2021; 69:51–57
10.
Leblond CS, Nava C, Polge A, et al: Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet 2014; 10:e1004580
11.
Escayg A, MacDonald BT, Meisler MH, et al: Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 2000; 24:343–345
12.
Saitsu H, Kato M, Mizuguchi T, et al: De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet 2008; 40:782–788
13.
Slager RE, Newton TL, Vlangos CN, et al: Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet 2003; 33:466–468
14.
Ng SB, Bigham AW, Buckingham KJ, et al: Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 2010; 42:790–793
15.
Kurotaki N, Imaizumi K, Harada N, et al: Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet 2002; 30:365–366
16.
Homsy J, Zaidi S, Shen Y, et al: De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015; 350:1262–1266
17.
Sanna-Cherchi S, Westland R, Ghiggeri GM, et al: Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest 2018; 128:4–15
18.
Flint J, Mackay TFC: Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res 2009; 19:723–733
19.
Sittig LJ, Carbonetto P, Engel KA, et al: Genetic background limits generalizability of genotype-phenotype relationships. Neuron 2016; 91:1253–1259
20.
Pizzo L, Jensen M, Polyak A, et al: Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet Med 2019; 21:816–825
21.
Davies RW, Fiksinski AM, Breetvelt EJ, et al: Using common genetic variation to examine phenotypic expression and risk prediction in 22q11.2 deletion syndrome. Nat Med 2020; 26:1912–1918
22.
Pounraja VK, Girirajan S: A general framework for identifying oligogenic combinations of rare variants in complex disorders. Genome Res 2022 32:904–915
23.
Harper AR, Nayee S, Topol EJ: Protective alleles and modifier variants in human health and disease. Nat Rev Genet 2015; 16:689–701
24.
Backman JD, Li AH, Marcketta A, et al: Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 2021; 599:628–634