Our understanding of genetic risk for neurodevelopmental disorders is based on studies of individuals clinically ascertained for specific diagnoses. While large-scale studies have identified hundreds of genes and rare variants for phenotypes such as autism, intellectual disability, and schizophrenia, a large fraction of the identified genetic variants are nonspecific and variable in both prevalence and degree of affectedness. Akin to the parable of the four blind men describing an elephant, studies on independently ascertained disease cohorts have often implicated the same genetic cause for distinct disorders. For example, mutations disrupting
SCN2A have been independently reported in cohorts manifesting epilepsy, schizophrenia, and autism (
1), and deletions on chromosome 15q13.3 have been associated with epilepsy, schizophrenia, and intellectual disability while further being documented in apparently unaffected individuals (
2). In fact, many disease-associated copy number variants—that is, deletions and duplications of genomic regions—have also been reported in unselected populations, where carriers of those variants were found to manifest subclinical psychiatric and cognitive defects (
3,
4). The variable expressivity of genomic variants that were originally believed to be highly penetrant toward a specific neurodevelopmental phenotype reveals that our current notion of disease pathogenicity is confounded by ascertainment bias of studying affected individuals from clinical cohorts.
In this issue of the
Journal, Shimelis and colleagues (
5) assess the impact of pathogenic mutations on neuropsychiatric outcomes in over 90,000 individuals from the Geisinger health care system population (termed the DiscovEHR cohort). Focusing on a curated list of 94 genes implicated in neurodevelopmental and psychiatric features, the authors analyzed exome sequencing data and found that 312 (0.34%) individuals carried a rare loss-of-function variant. The observed prevalence of pathogenic mutations was several times lower than prevalences reported in disease cohorts (
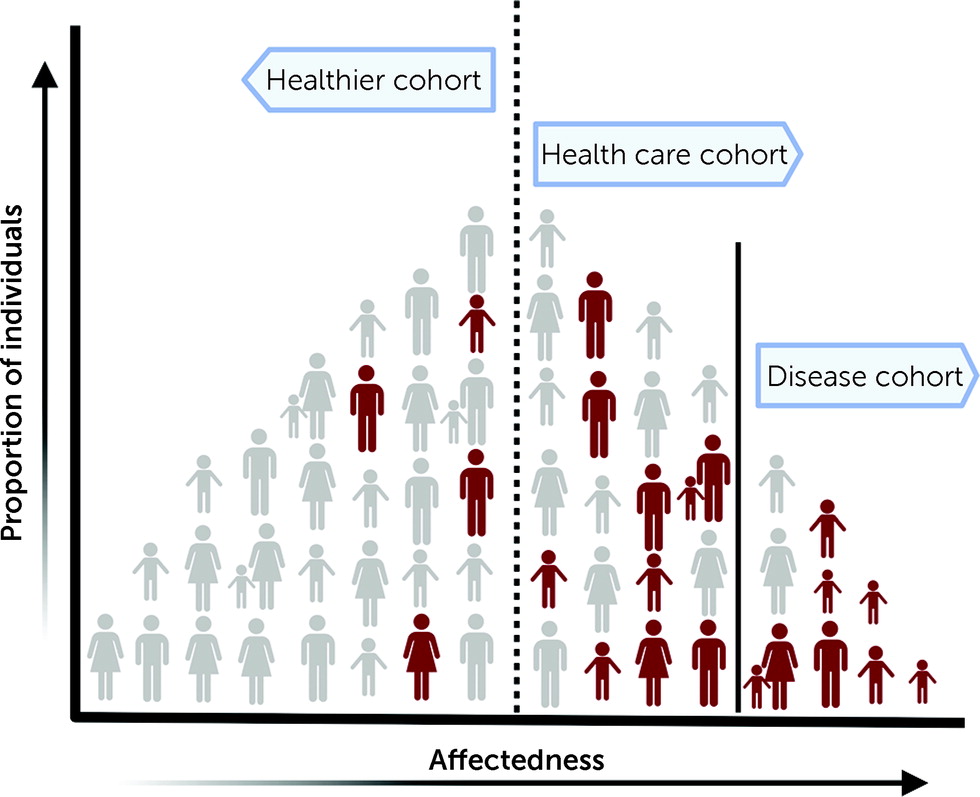
6). Although more representative of the general population than a disease-specific cohort, cohorts derived from health care systems may overrepresent individuals with a clinical diagnosis and hence show a higher prevalence of pathogenic variants than an unselected general population (
Figure 1). In fact, the prevalence of pathogenic copy number variants in the DiscovEHR cohort was higher compared to that of individuals from the general Icelandic population (
4), healthy aging adults in the UK Biobank (
7), or participants in the Estonian Biobank studies (
8). While survivor bias, a type of selection bias arising from healthy volunteers participating in cohorts such as the UK Biobank, could account for some of these differences, prevalence estimates in unselected populations could inform us on the selective effects of rare variants and their disease relevance in a clinical context.
To assess the penetrance of the pathogenic variants, the authors identified individuals with variants who also had an ICD-10 diagnosis for 14 neurodevelopmental and psychiatric features. Only 34% of individuals with a pathogenic variant had an ICD-10 diagnosis. This is noteworthy, as the 94 genes selected in this study are mostly known to be associated with highly penetrant disorders, typically diagnosed in early childhood. The authors identified pathogenic mutations in 61 of the 94 genes, including those associated with a range of neuropsychiatric outcomes, such as
ANK2 (
9) and
SHANK2 (
10). Some genes recapitulated known associations, such as
SCN1A and
STXBP1 for epilepsy (
11,
12), while other genes showed novel associations with previously unreported phenotypes. For example, epilepsy in an individual with a mutation in
RAI1, the causative gene for Smith-Magenis syndrome (
13), and depressive disorder in individuals with mutations in
KMT2D or
NSD1, genes associated with Kabuki syndrome (
14) and Sotos syndrome (
15), respectively, were noted. Some pathogenic variants may also show pleiotropic effects, with individuals manifesting features outside cognitive and behavioral domains, such as cardiac or renal defects, due to shared genetic etiologies (
16,
17). Indeed, Shimelis et al. found that about 11% of individuals with pathogenic variants had congenital anomalies, with 5.4% manifesting cardiac or renal disorders. When the authors included the diagnosis of anxiety and depression or a history of congenital malformation in their analysis, their overall penetrance estimates increased to 68.6% and 71.2%, respectively. These observations indicate that genes that typically cause syndromic diagnoses in affected children can be associated with milder neuropsychiatric or unexpected pleiotropic features in a broadly ascertained adult population.
Although ICD-based diagnosis may not capture the entire spectrum of phenotypic effects, the substantially reduced penetrance of pathogenic variants detected in this study has significant implications for understanding the complexity of genetic disorders. The role of the genetic background, which is a collection of variants of different classes and frequencies that co-occur with the primary pathogenic variant, in modulating disease risk across ascertainments cannot be overstated. The effects of naturally occurring genetic variants on behavioral traits have been well documented in tractable model systems, such as mice and flies (
18). For example, Sittig et al. (
19) found significant variation in behavioral phenotypes when mice lacking bipolar-associated genes were tested under different strain-specific genetic backgrounds. A multi-hit model has been proposed and explored in the context of variably expressive variants in humans, in which a primary variant sensitizes the genome toward risk for a range of neuropsychiatric outcomes and its interplay with other variants determines the trajectories toward distinct disorders (
20). For example, Davies et al. (
21) found a substantial contribution of schizophrenia polygenic risk scores toward both cognitive defects and schizophrenia among individuals with 22q11.2 deletion. Another implication of low-penetrance variants is their high likelihood of being transmitted across generations. These variants may cause mild features, as observed in this study, but in conjunction with other rare variants may cause more severe disease in subsequent generations. For example, a recent study showed that individuals having pathogenic mutations in two or three genes were more likely to manifest intellectual disability than those with mutations in individual genes (
22). While these models explain increased disease risk, the reduced penetrance of pathogenic mutations observed by Shimelis et al. could in part be due to a protective effect of other variants in the genetic background that may alleviate risk for disease (
23). For example, Backman et al. (
24) found that a missense variant in
ST6GALNAC5 within the UK Biobank cohort was associated with protection against lower gray–white matter contrast, a feature linked with an increased rate of cognitive decay. Protective alleles are generally harder to find because of their rarity, but population-scale genetic studies show promise in this area and could increase the list of potential therapeutic targets relevant to disease. As illustrated by the Shimelis et al. study, analyzing populations not selected for a specific disorder has enabled reevaluation of disease gene pathogenicity within the context of the genetic background.
Currently, a majority of the efforts to understand disease etiology are concentrated on identifying high-effect-size rare variants and individual or collective effects of common variants, but it is becoming increasingly clear that disease penetrance is a result of an interplay between pathogenic mutations and the genetic background. Population-based biobanks provide an unbiased representation of the entire spectrum of genomic variation and increased power to identify genetic associations for complex disorders. Insights derived from population-based studies of neuropsychiatric disorders will allow for a genotype-first approach to diagnosis, timely intervention due to better understanding of disease prognosis, and identification of drug targets and treatment options owing to an improved perception of disease mechanisms.