Mental disorders are among the most heritable complex human disorders. For schizophrenia, bipolar disorder, depression, and attention deficit hyperactivity disorder (ADHD), twin studies have estimated broad sense heritabilities of 40%–80% (
1–
4). Genome-wide association studies (GWASs) have since shown that approximately one-third is driven by thousands of common genetic variants, each with individually small effect (
5). As ever-expanding GWAS sample sizes bring genetic prediction and stratification tools closer to the clinic (
6), a better understanding of the complex genetic architecture of mental disorders is needed to address questions regarding disease classification and the potential for precision medicine in psychiatry.
Larger GWAS sample sizes have also revealed extensive shared genetic risk variants across diagnostic categories, mirroring their overlapping clinical characteristics (
7). A meta-analysis of eight mental disorders identified 109 independent genetic loci associated with two or more disorders (
8). Interestingly, 11 of these had “discordant” effects, that is, they increased the risk of one disorder but decreased the risk of a second (
8). This is supported by findings from pairwise analyses (
9,
10) that have identified hundreds of shared loci between mental disorders and related traits such as intelligence (
11,
12) and personality traits (
13), with a mixture of concordant and discordant effects (
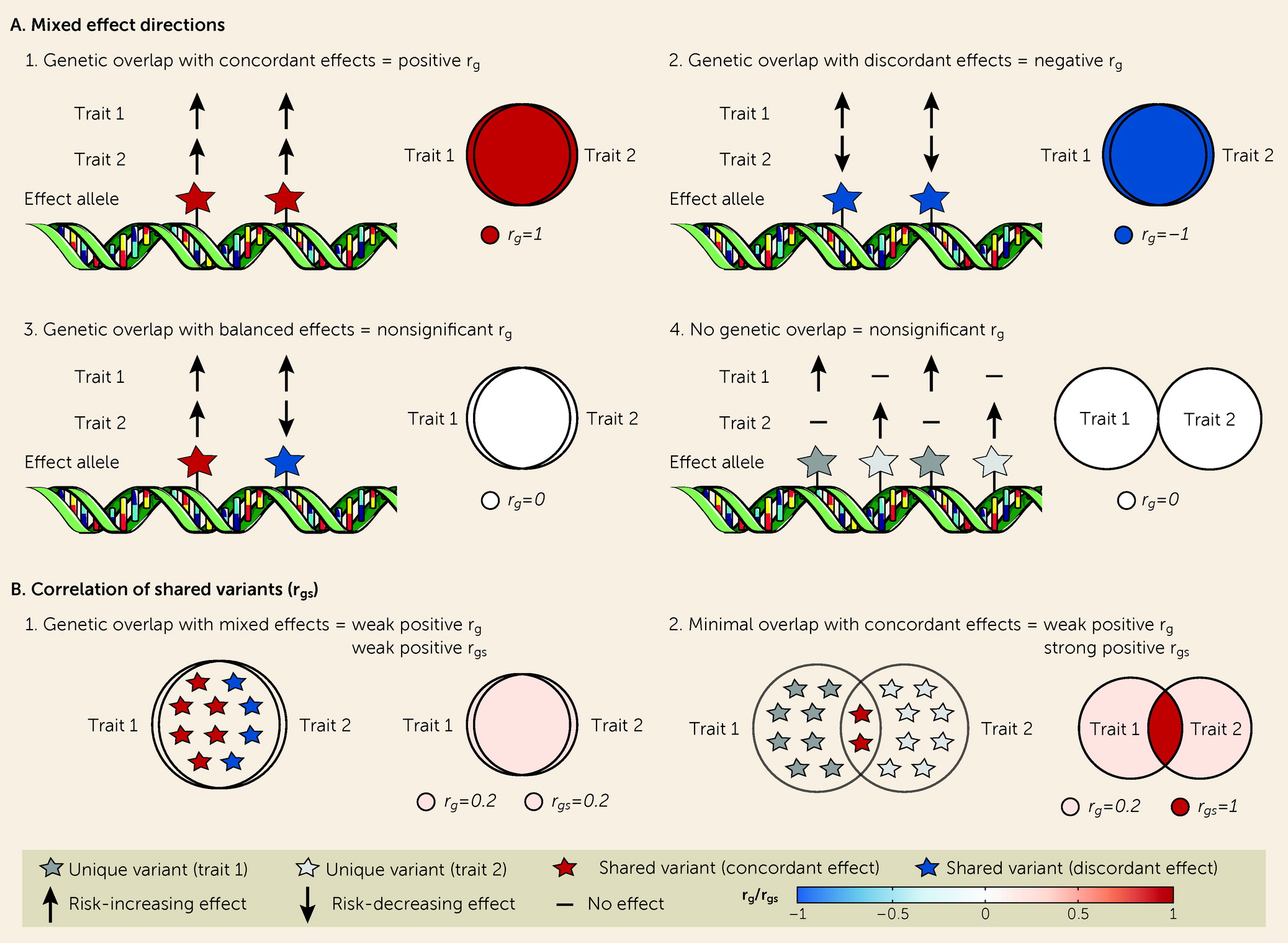
Figure 1A). Such extensive “pleiotropy” calls into question the traditional conceptualization of genetic risk, in which a specific set of genes is implicated for a specific disorder (
14).
Although r
g is an informative measure of the genetic similarity between two phenotypes, it does not capture all dimensions of genetic overlap. Shared genetic variants can have either concordant or discordant effects (
12,
17,
18). However, since r
g is a genome-wide summary measure, it does not differentiate genetic overlap with a mixture of concordant and discordant effects from an absence of genetic overlap, returning an estimate close to 0 in both scenarios (
Figure 1A). It is necessary to capture this “missing dimension” of genetic overlap to comprehensively describe the shared genetic underpinnings of polygenic mental disorders (
17). Overlap despite minimal genetic correlation may indicate shared molecular mechanisms, with implications for how we conceptualize genetic risk for mental disorders.
The bivariate causal mixture model (MiXeR) (
19) and LAVA (
20) can shed light on this “missing dimension.” MiXeR circumvents the need to identify all “causal” variants by inferring the total number of “causal” variants for each trait (univariate) and the total number of shared and unique “causal” variants for a pair of traits (bivariate) (
19). MiXeR also estimates the genetic correlation of shared variants (r
gs), in addition to r
g (
Figure 1B). Using MiXeR, we have demonstrated extensive genetic overlap across several mental disorders and related traits with mixed effect directions (
13,
18,
21). The relevance of mixed effects has been further emphasized by LAVA, which calculates local genetic correlations across the genome (
20). Despite employing a distinct statistical framework, LAVA revealed widespread local genetic correlations across somatic and mental traits with mixed effect directions, even in the presence of minimal r
g (
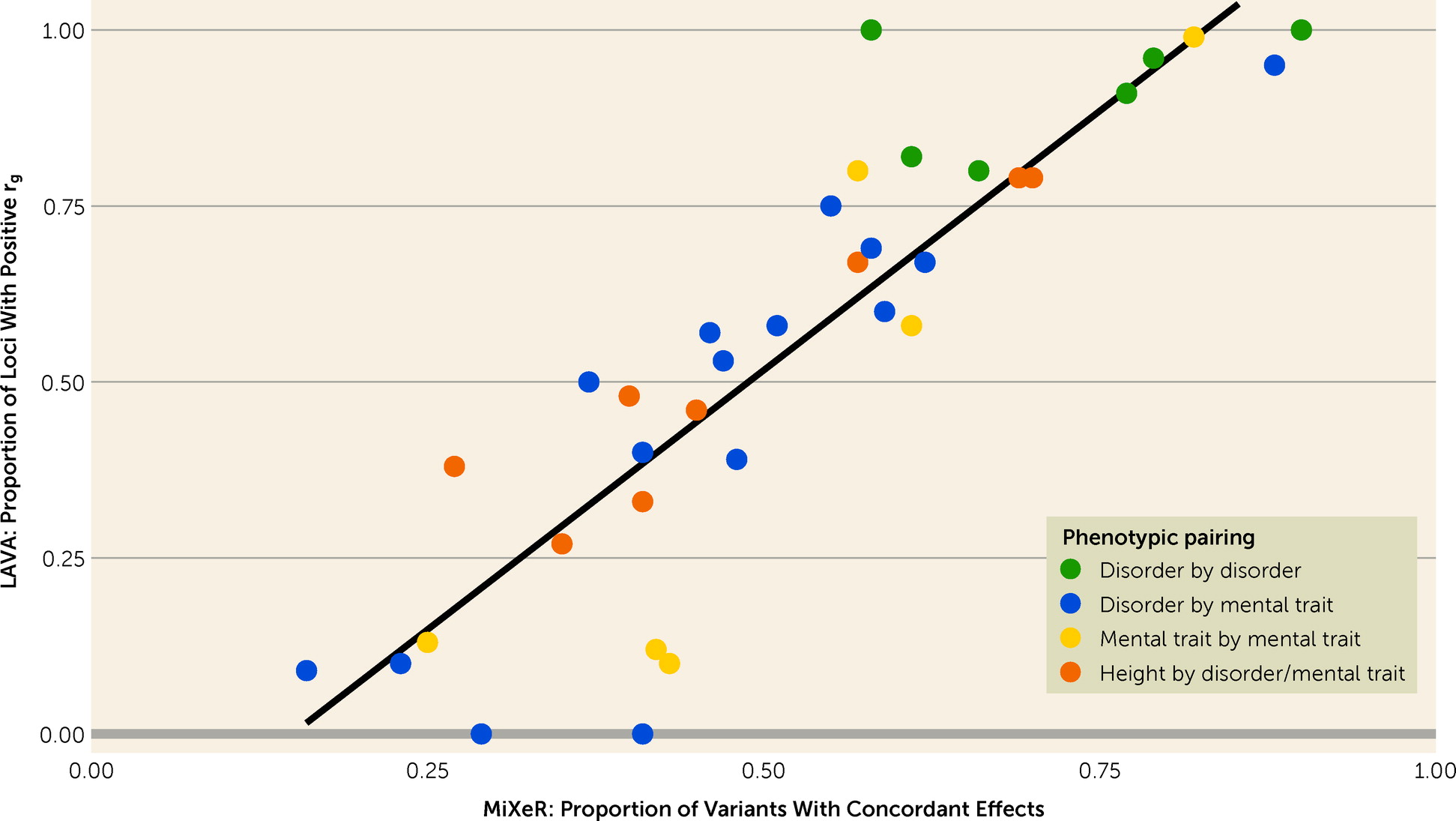
20). However, neither MiXeR nor LAVA has been systematically applied across mental disorders and cognitive and personality traits using the largest-to-date GWASs.
We applied MiXeR and LAVA to large-scale GWASs of ADHD, bipolar disorder, schizophrenia, depression, and cognitive and personality traits to characterize their genetic overlap beyond rg. We aimed to map 1) polygenicity, 2) genetic overlap allowing mixed effect directions, 3) rgs, and 4) local genetic correlations, using height as a comparator. We compared MiXeR estimates of mixed effect directions to LAVA local correlations to validate our findings across two distinct statistical frameworks. By charting the landscape of genetic overlap beyond rg, we provide insights into the unique and shared genetic architectures underlying psychiatric disorders, with implications for how we conceptualize genetic risk for mental disorders and related traits.
Discussion
In this cross-trait genetic analysis of ADHD, bipolar disorder, depression, schizophrenia, and cognitive and personality traits, we systematically quantified genetic architecture beyond genome-wide genetic correlations. We found marked differences in polygenicity but extensive genetic overlap across all mental disorders and cognitive and personality traits, with few disorder-specific variants. These findings were supported by LAVA local correlations, which also revealed patterns of mixed effect directions concealed by estimates of genome-wide genetic correlations. This indicates that, rather than a predominance of disorder-specific risk variants, there may be a set of highly pleiotropic variants that influence the risk of diverse mental disorders and related traits. By extension, phenotypic specificity may be largely driven by the distribution of effect sizes and effect directions across this pool of pleiotropic variants rather than variants unique to each phenotype (
17). Building on previous work highlighting extensive overlap across mental disorders (
13,
17), this represents a conceptual advance in our understanding of the genetic architecture of mental disorders, which may inform strategies for genetic discovery, biological characterization, and psychiatric nosology, providing the foundations for the development of precision psychiatry and treatment stratification across diagnostic boundaries.
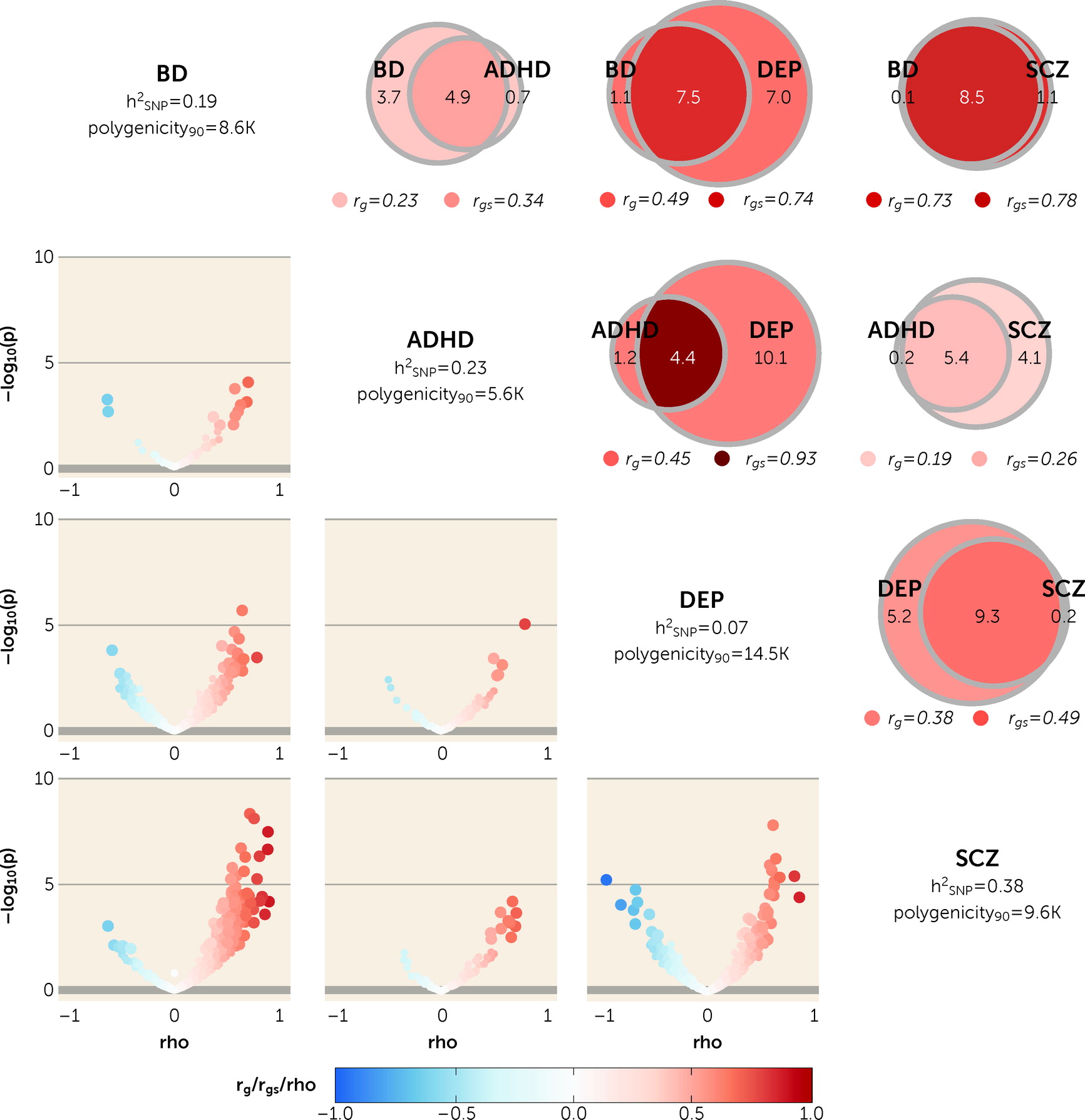
First, we used univariate MiXeR to show that the genetic architectures of mental traits exhibit fundamental differences beyond heritability, with differences in polygenicity across the eight included traits. Among mental disorders, this was most pronounced between ADHD and depression, although lower polygenicities have recently been reported for migraine (
23), cortical MRI measures, and other somatic traits (
23–
25). While the neurobiological and clinical implications of these findings are currently speculative, it is possible that polygenicity is a marker of heterogeneity at the neurobiological and/or clinical level. For example, ADHD may represent a more neurobiologically and/or clinically homogeneous population than depression. By extension, polygenicity may be a useful marker of genetic heterogeneity, which could be used to test the effect of biomarkers or clinically defined subgroups on the genetic makeup of a given disorder. Differences in polygenicity may also be due to differences in biological complexity or negative selection (
26,
27). More deeply phenotyped samples and improved functional characterization of genetic loci are required to provide further insights.
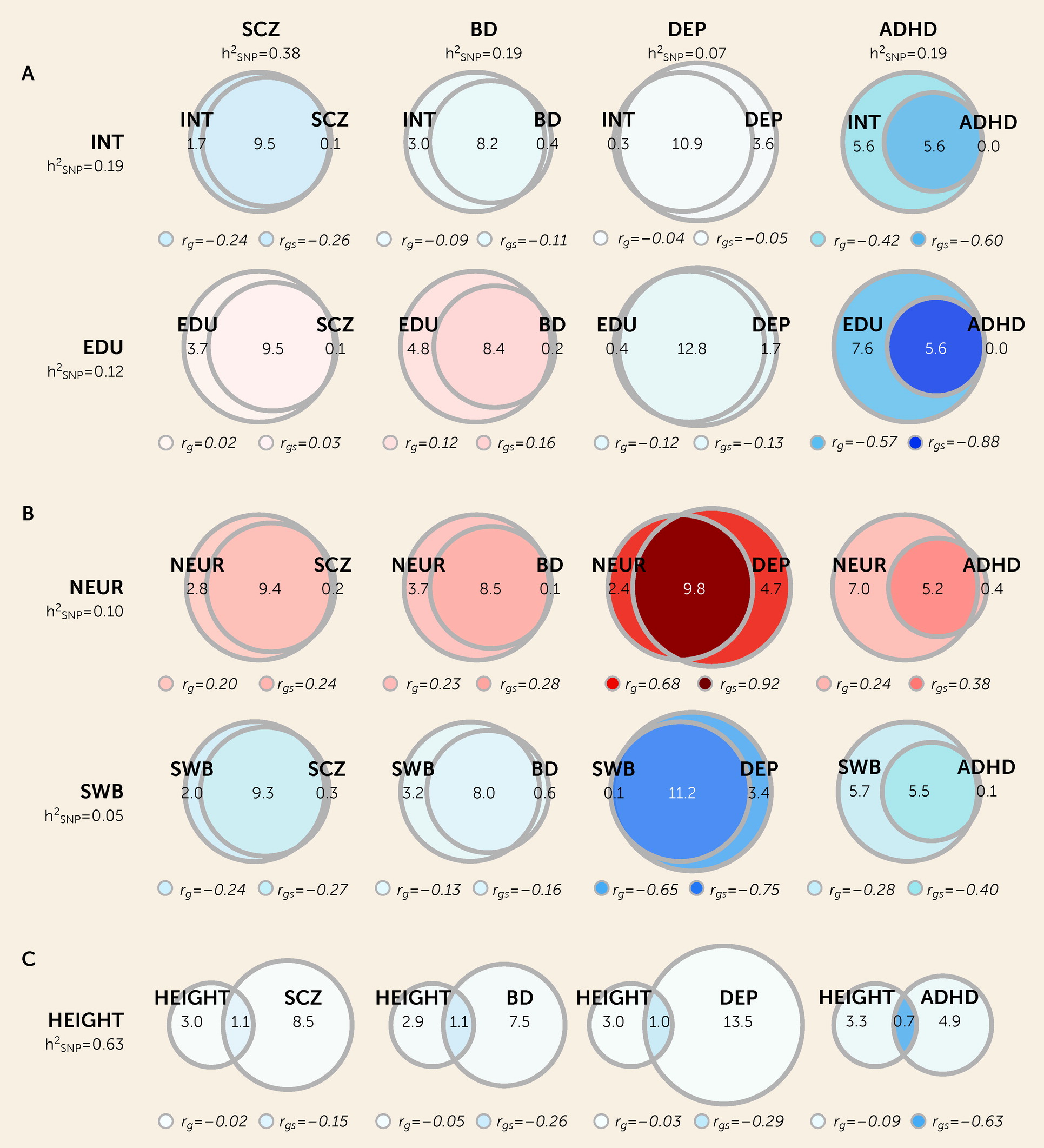
Using bivariate MiXeR, we found extensive genetic overlap across mental disorders and cognitive and personality traits. This pattern was present in scenarios of weak genome-wide genetic correlation, such as depression and cognition, as well as strong genome-wide correlations, such as bipolar disorder and schizophrenia. The former is indicative of a balance of shared variants with concordant and discordant effect directions on each trait, a pattern we replicate using LAVA. These findings build on previous evidence demonstrating extensive genetic overlap between schizophrenia, bipolar disorder, depression, and intelligence, with widespread mixed effect directions (
11,
17). Together, this indicates that most common variants that influence the genetic risk for diverse mental phenotypes are highly pleiotropic and may have both risk-enhancing and risk-reducing effects on different disorders and traits. Consequently, it may be the specific distribution of effect sizes of highly pleiotropic variants that predominantly contributes to the development of a given mental disorder rather than a set of phenotype-specific variants.
This conceptual insight extends the current theories of genetic susceptibility for mental traits and disorders. These are traditionally based on the assumption that a specific set of genes is implicated for a given mental disorder, with varying degrees of genetic overlap across disorders, reflected by their genetic correlations (
8,
16,
28). However, this is inconsistent with the extent of genetic overlap observed when accounting for mixed effect directions, which may provide a better conceptual framework for the neurobiology of mental traits and disorders. The brain is a complex organ with abundant pleiotropy across different brain regions (
29), brain functions (
30), and mental traits (
31). Thus, it is likely that differences in activity in the same neurobiological systems alter diverse mental traits and disorders, but the magnitude and direction of effect may differ across and within diagnostic categories, exemplified by evidence of increased glutamatergic neurotransmission in schizophrenia (
32) but decreased transmission in depression (
33). This is also consistent with the fact that most genetic variants associated with mental disorders reside within regulatory elements rather than coding regions (
34). Allelic variation may therefore “tune” neurobiological pathways in different directions, resulting in phenotypic differences mediated by the same pathways (
35). By applying statistical tools that allow for mixed effect directions and estimation of effect sizes, new insight into neurobiological substrates for mental disorders is possible.
These findings also have clinical implications. First, evidence of extensive genetic overlap with limited trait specificity underscores the limited extent to which our current categorical diagnostic system maps onto underlying biological processes (
36). These findings may therefore be more consistent with dimensional approaches to psychiatric nosology, which allow for specific combinations of symptoms as well as interactions with other mental traits, as proposed by the Research Domain Criteria (RDoC) or Hierarchical Taxonomy of Psychopathology (HiTOP) frameworks (
37,
38). This may also help to explain the large degree of comorbidity and the prominence of overlapping clinical characteristics observed across mental disorders. Alternatively, once greater proportions of the SNP-based heritability of mental disorders have been characterized, it may be possible to parse the heritable component into constituent biological processes. This may enable construction of a personalized, biologically informed diagnostic system, similar to the “palette model” proposed for diabetes (
39). As the era of large-scale case-control GWASs transitions toward deeply phenotyped clinical and population samples (
40), it will be of great interest whether subphenotyping results in more specific genetic signals. This will also be highly relevant for the clinical application of polygenic risk scores, which require not only improved explained variance but also the ability to discriminate across diagnostic groups or clinically relevant decisions.
Despite the pervasive overlap, the number of shared and unique “causal” variants varied across phenotypic pairs, sometimes revealing the presence of strongly correlated shared variants despite moderate genome-wide genetic correlation. This pattern was most evident between ADHD and depression, a finding supported by LAVA. This suggests that although a large proportion of depression-associated variants are not associated with ADHD, those that are shared have highly similar effects. This mirrors recent findings from a genomic structural equation modeling approach that clustered depression with neurodevelopmental disorders, including ADHD (
8). This may indicate distinct clinical subgroups with higher rates of comorbidity, or shared involvement of specific molecular mechanisms with similar effects on both disorders. Further investigation of the shared genetic architecture of ADHD and depression may prove fruitful for the identification of underlying biological mechanisms and new treatment targets (
41–
43).
There was substantially less genetic overlap between mental disorders and height, with approximately 1,000 shared “causal” variants across all pairwise analyses. This may represent nonspecific “background” genetic overlap observed across all polygenic traits, or, alternatively, it may capture highly pleiotropic transcription factors or regulatory elements that influence diverse traits across distinct tissues and systems. (See the Supplementary Discussion section in the
online supplement for additional discussion of the relevance of our findings to the
p factor hypothesis.)
This study had limitations. Only samples of European ancestry and common genetic variants were included, because of limited availability of trans-ancestral and sequenced samples. Both educational attainment and subjective well-being are influenced by social factors, and neither can be considered direct measures of cognition or personality. Nonetheless, there is a lack of well-powered GWASs of more specific measures within either domain, and these measures provide useful insights alongside intelligence and neuroticism. We also cannot exclude the possibility of comorbidity or misdiagnosis across psychiatric samples. However, the extent of the overlap observed and its consistency across mental traits indicate that this alone cannot explain our findings. In addition, for all pairwise analyses of mental disorders and related traits, besides depression and neuroticism, the AIC differences were negative when compared to maximum possible overlap. This indicates that MiXeR-modeled estimates of genetic overlap are indistinguishable from maximum overlap as measured by AIC, and so must be interpreted with caution. It is important to note, however, that the overlaps presented are still the best-fitting estimates as determined by maximum likelihood estimation. Finally, given the need to maximize sample sizes, it was not possible to perform replication analyses in independent samples. Nonetheless, we used both MiXeR and LAVA to triangulate our findings using different statistical frameworks. Our univariate measures are further supported by recent findings using Fourier mixture regression (
44), which predicts required sample sizes for a given proportion of explained heritability, like MiXeR (see the
online supplement). For schizophrenia, bipolar disorder, and neuroticism, which were common to both analyses, there was high concordance of predicted sample sizes required to explain 90% SNP heritability.
In summary, we have used advanced statistical modeling to demonstrate both considerable similarities yet also fundamental differences in the genetic architecture of ADHD, bipolar disorder, schizophrenia, and depression, alongside cognitive and personality traits. Despite extensive genetic overlap and few trait-specific variants, there were distinct patterns of genetic correlations with widespread mixed effect directions. This suggests that it is the specific distribution of effect sizes of highly pleiotropic variants that predominantly contribute to the development of mental disorders and related traits, rather than a set of disorder-specific variants. This represents a conceptual advance in our understanding of the genetic risk of mental disorders, suggesting that normative and pathological mental traits, and the biological processes underlying them, exist on the same dimensions in genomic space. These findings place greater emphasis on efforts to improve the specificity of psychiatric diagnostic categories, potentially offering a means to test the genetic heterogeneity of hypothesized subgroups through estimates of polygenicity. This may aid efforts to refine the current nosological system, with potential for improved translation of genetic findings into clinically meaningful prediction and stratification tools, and improved drug target identification.
Acknowledgments
The authors thank the research participants, the employees, and the researchers of the Schizophrenia, Depression, Bipolar Disorder, and ADHD Working Groups of the Psychiatric Genomics Consortium, 23andMe, UK Biobank, CTG, SSGAC, and GIANT for making this research possible. This work was partly performed on the TSD (Tjeneste for Sensitive Data) facilities, owned by the University of Oslo and operated and developed by the TSD service group at the University of Oslo, IT Department (USIT). Computations were also performed on resources provided by UNINETT Sigma2, the National Infrastructure for High-Performance Computing and Data Storage in Norway. The authors gratefully acknowledge support from NIH (NS057198, EB000790, 1R01MH124839), the Research Council of Norway (229129, 213837, 223273, 324252, 248980), the South-East Norway Regional Health Authority (2017-112, 2019-108), and KG Jebsen Stiftelsen (SKGJ-MED-021). This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement nos. 847776, 964874, and 801133 (Marie Skłodowska-Curie grant agreement).