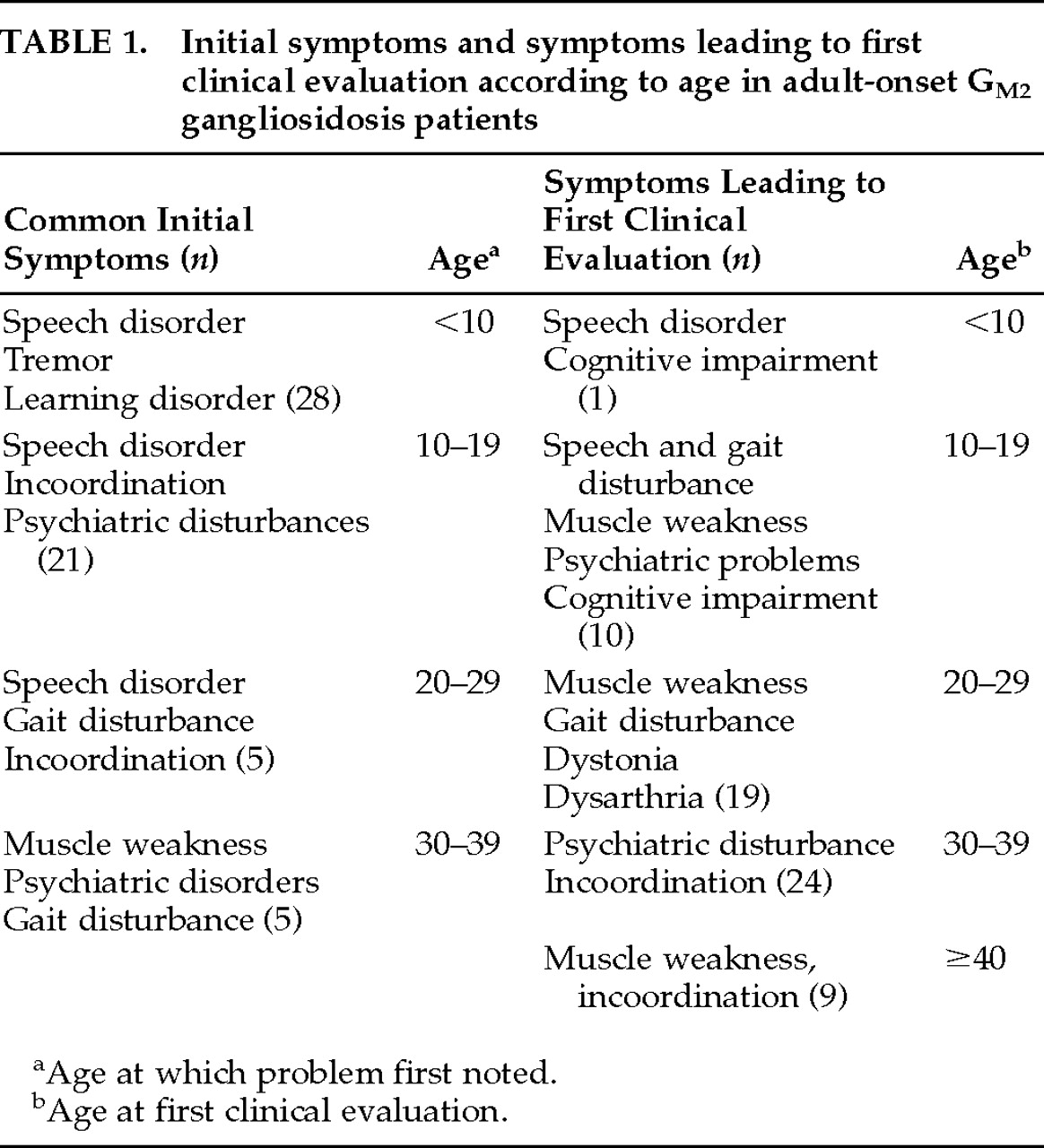
Tay-Sachs disease (TSD) is an autosomal, recessively inherited lipid storage disease in which gangliosides accumulate within neurons as a result of a deficiency of the enzyme β-hexosaminidase A (HEX A). It is now clear that this disorder encompasses a very wide age and clinical spectrum.
1,2 The classic or infantile form
3,4 is a rapidly progressive neurodegenerative disorder characterized by motor weakness, myoclonic jerking, loss of developmental milestones, a characteristic macular cherry-red spot, and death within the first few years of life. A late infantile form of HEX A deficiency (also called juvenile onset)
5–7 has been described, with the onset ranging from 1 to 9 years. There are no typical neonatal problems, and early milestones are often normally achieved. Neurological deficits then develop and are prominent. They commonly include seizures, choreiform and dystonic movement disorders, incontinence, speech problems leading to complete mutism, dysphagia, and severe gait disturbances with eventual immobility. Dementia appears to supervene in all cases, and death occurs by age 15.
More recently, a G
M2 gangliosidosis variant designated “adult onset”(AGG),
8,9 has been described in which both psychiatric and neurologic disorders occur. Afflicted individuals often exhibit mild or no symptoms until young adulthood. Although the most common features include disturbances of speech and coordination, psychosis and mood disturbances can be the presenting features, so that a psychiatrist may be the first health professional to assess such a patient. The age of onset can be difficult to determine in the adult variant. Even when one can date signs and symptoms back to childhood, the more benign course of illness leads to its being classified as adult onset. In the following, we review the etiology and pathophysiology of AGG. We then summarize a literature review of 64 cases of AGG, describing the neuropsychiatric symptoms and treatment options.
GENETICS OF AGG
The major isoenzymes of hexosaminidase in humans are HEX A and HEX B. HEX A is a trimer composed of alpha and beta chains; HEX B is a homopolymer composed of beta subunits. A mutation on chromosome 15 in the alpha chain region will result in a selective deficiency of HEX A. A mutation on chromosome 5, which alters the region coding for the beta subunit, will affect both HEX A and HEX B, producing what is termed the Sandhoff variant of the disease, characterized by visceral organ involvement as well as neuropsychiatric disturbances.
The infantile form of Tay-Sachs is associated with severe mutations on chromosome 15 resulting in formation of no, or highly unstable, mRNA and thus a total absence of the protein (alpha subunit) coded by that mRNA. In the adult variant of the illness, a glycine-serine point mutation within the protein coding regions leads to stable mRNA and production of the corresponding protein subunits, but the subunits are defective.
10–13 Patients with juvenile forms of the illness have point mutations at a different site on the gene than that associated with the adult form, suggesting that the clinical division of the G
M2 gangliosidoses into infantile, juvenile, and adult forms may correspond to variable mutation sites within the gene. It has been proposed that within the realm of the adult variant specific mutations may be correlated with certain symptom patterns, but this has yet to be empirically confirmed.
1,14PATHOGENESIS OF TSD AND VARIANTS
Gangliosides are sialic acid-containing glycophospholipids that are integral components of all plasma membranes. In the brain they are found predominantly in neurons, although they are also present in glia and myelin.
15 Gangliosides are the major lipid components of neuronal plasma membranes and are composed of a lipophilic portion, which is embedded in the membrane, and a hydrophilic portion, which protrudes onto the surface. They are thought to be involved in neuritogenesis, synaptogenesis, neuronal differentiation and regeneration, and cell-to-cell interactions, and to act as receptors for certain hormones and bacterial toxins.
15–17In the normal process of catabolism, gangliosides are removed from the membrane by an activator protein with which it forms a complex. This ganglioside–protein activator complex constitutes the substrate that is transported to the lysosome within the cell body and degraded, in the case of the G
M2 ganglioside, by HEX A and HEX B. Inability to process the ganglioside may result from an abnormality of either the isoenzyme or an activator protein. There is some evidence of a correlation between the level of enzyme activity and the severity of the disease.
18It appears that neuronal function is compromised in AGG as the residual enzyme ultimately falls behind in the processing of the gangliosides and unmetabolized substrate accumulates within lysosomes.
18,19 These depositions are not believed to be directly cytotoxic, and they are present long before there is any evidence of cell death. In some neurons the storage materials simply increase cell size, whereas in others they give rise to unique morphological abnormalities. In 1976 Purpura and Suzuki
20 described outgrowths in the region between the cell body and the axon of cortical pyramidal neurons taken from a brain biopsy of a 14-month-old child with G
M2 gangliosidosis. These “meganeurites“
20 were associated with aberrant dendritic, neuritic, and synaptic growth. Purpura studied these morphological changes further in mutant cat models of G
M1 and G
M2 gangliosidoses. He noted the selectivity of these changes for certain neuronal populations, particularly the medium spiny neurons of the caudate nucleus and the small and medium cortical pyramidal neurons.
21–24 The relationship between the excessive abnormal neuronal connectivity and the clinical nature of the disease remains to be elucidated.
Siegel and Walkley
25 have recently demonstrated a correlation between the amount of G
M2 ganglioside accumulation and the extent of ectopic dendritic growth in cortical pyramidal neurons in a number of neuronal storage diseases. These data suggest that abnormalities in the processing of G
M2 gangliosides may be a final common pathway for degeneration, regardless of the primary metabolic defect. The finding that gangliosides are potent inhibitors of protein kinase C,
26 important in the transduction of neurotransmitter signals, suggests another possible link between the abnormal accumulation of gangliosides and neuronal dysfunction. More recent studies have shown that in a number of storage diseases, including the gangliosidoses, GABAergic neurons develop axonal enlargements termed
spheroids.
23Overall, it is not clear why neurons are targeted in the absence of hexosaminidase and, beyond that, why subpopulations of neurons are characteristically (but variably) affected. Several potentially important factors include higher production rates of substrate in certain cells, different levels of residual enzyme in specific cell populations, and differential ability of the mutated residual enzyme to function within variable intracellular environments.
19 Which, if any, of these factors actually contribute to the observed pattern of symptoms in the gangliosidoses remains unclear.
FAMILIAL INVOLVEMENT
As discussed above, AGG is transmitted by an autosomal recessive pattern of inheritance. Relatives of affected individuals may therefore be normal, they may be carriers, or they may have the illness. Beyond the recognition that the carrier states tend to have intermediate levels of hexosaminidase, little attention had been given to the potential for phenotypic expression of the carrier state. Several reports have included hexosaminidase levels of patients' families,
44,45 but reports of clinical findings in these people are lacking.
Hexosaminidase levels as well as clinical and neurophysiological changes have been reported for 14 carriers in a family with a beta subunit mutation.
50 Upper and lower motor neuron signs, as well as psychiatric manifestations (depression, anxiety, behavioral changes) were present to a variable degree in 13 of these heterozygotes. Although we recognize the difficulty in quantifying “behavioral change”or severity of “cramps”for a numerical analysis, there appeared to be some relationship between hexosaminidase level and extent of symptoms. We assigned point values to the symptoms described by Federico et al.,
50 based on the described severity of the symptoms. We then totaled the symptoms present for each carrier that had a hexosaminidase level provided (
n=13) and did a correlational analysis plotting hexosaminidase level against symptom totals. There was a significant correlation between hexosaminidase level and expression of symptoms in the carrier state (
n=13,
r=0.75,
r2=0.56,
t=5.5 where
t[df=12, α=0.01]=2.68). It is well recognized that females who are heterozygotes for inherited illnesses such as adrenoleukodystrophy and fragile X syndrome can develop clinical syndromes that range from mild to severe neuropsychiatric impairment.
52–55 It appears that phenotypic expression of the heterozygous state occurs in AGG as well; it is thus important that all family members of AGG patients receive a thorough neuropsychiatric examination in addition to measurement of hexosaminidase levels.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of AGG is extensive because of the variability in clinical presentation. In any patient with psychiatric illness, the presence of neurologic symptoms or cognitive impairment should raise the index of suspicion and lead to further evaluation.
49,52 Several storage diseases are likely under-recognized as illnesses that may present in adulthood with psychiatric features. In addition to AGG, as outlined here, X-linked adrenoleukodystrophy,
56 metachromatic leukodystrophy,
57–63 ceroid neuronal lipofuscinosis,
63–66 hepatolenticular degeneration,
67–69 Niemann-Pick type C,
70,71 and cerebrotendinous xanthomatosis
72,73 should be included in the differential when unusual or refractory neuropsychiatric symptom clusters are present.
There are no pathognomonic physical findings associated with AGG. Neither the characteristic macular cherry-red spot apparent in the infantile form of Tay-Sachs nor the visceral organ involvement seen in infantile Sandhoff's disease are present in the adult variants of these diseases. Dysmorphism, characteristic of the GM1 gangliosidoses, is not found in the GM2 gangliosidoses. The neurological signs and symptoms outlined above may or may not be present in the early stages of the disease.
Typical screening investigations will usually provide no evidence of an underlying metabolic defect such as AGG. Although there are reports of elevated lactate dehydrogenase and creatinine phosphokinase in AGG patients,
1,31 these are nonspecific findings, and the majority of case reports describe normal serum chemistries. Similarly, EEGs are commonly normal. Cranial CT and MRI studies often show cerebellar atrophy, although they too can be normal.
38,49 In a recent study, no correlation between clinical signs and radiologic changes (CT and MRI) in AGG was demonstrated.
74 We were similarly unable to demonstrate a correlation between the presence of cerebellar signs in the clinical summary and the reports of cerebellar atrophy in the cases reviewed.
Electromyographic (EMG) studies were abnormal in 89% of the 35 patients for whom results were reported. Although Mitsumoto et al.
36 have claimed to find a unique pattern of complex repetitive discharges in their AGG patients, there was no characteristic abnormality reliably documented in association with AGG in other EMG studies.
Thus, although imaging and EMG studies may document the presence of an underlying neurologic process, there are no findings in standard investigations to suggest a diagnosis of AGG. It is important, therefore, to order hexosaminidase levels from serum, leukocytes, or cultured skin fibroblasts to diagnose AGG when it is suspected. Rectal biopsy may confirm the presence of myenteric plexus neurons swollen with cytoplasmic membrane bodies, but this is not a necessary test for diagnostic confirmation.
TREATMENT
There is no specific treatment for AGG. Neurologic sequelae are managed by supportive measures, along with orthopedic and rehabilitative measures where indicated.
Treatment of the psychiatric manifestations of AGG is controversial. Although Rubin et al.
43 reported successful treatment of an AGG patient in an acute paranoid state, most reports suggest that neuroleptic medications are rarely efficacious. Streifler et al.
45 described 3 psychotic patients with AGG who demonstrated poor response to psychoactive medication. Hurowitz et al.
48 reported no benefit from medications in 2 other patients with prominent mood disorders, and Renshaw et al.
47 noted little effectiveness of traditional antipsychotics and antidepressants in an individual with psychosis and depression. Our patient
49 received a long trial of high-dose neuroleptic medication (mean daily dose 1,000 mg in chlorpromazine equivalents) with no benefit. Thus, from the data available it would seem that the efficacy of neuroleptics in these patients is limited.
A further concern regarding the use of antipsychotic medication in AGG patients comes from reports that amphiphilic drugs, including phenothiazines and tricyclic antidepressants, increase lipidosis.
75,76 It has been reported that patients with psychiatric symptoms and AGG have a more severe illness course, with the assumption being that treatment with these drugs worsened the underlying illness. While this theoretical concern is valid, there may be other ways to account for the observed worsening of symptoms in AGG patients treated with neuroleptic medication. Drugs such as neuroleptics can produce many of the extrapyramidal symptoms commonly described in AGG patients independent of worsening of the underlying disease process, and the anticholinergic properties of neuroleptics can cause sedation and cognitive impairment. Thus, a patient with AGG on phenothiazines might appear more neurologically impaired, but this may represent drug side effects rather than acceleration of the illness.
Individuals with a variety of brain lesions or abnormalities are, in general, believed to be more sensitive to psychoactive medication. The presence of movement disorders in untreated patients with AGG implicates the basal ganglia and suggests that these patients might be especially vulnerable to the extrapyramidal complications associated with the use of neuroleptics. Streifler et al.
45 noted that attempts to treat the psychosis of an AGG patient resulted in “severe adverse reactions, mainly of extrapyramidal type” and eventually led to a catatonic state that resolved only after the medication was discontinued. Thus, although AGG patients may well be especially vulnerable to the effects of neuroleptic drugs, there are explanations for this sensitivity that do not require an actual effect of the drug on the underlying disease process.
In order to demonstrate such an effect, one would have to have evidence of prolonged impairment of a kind not typically produced by neuroleptics and persisting after neuroleptics are discontinued. A worsening of symptoms such as ataxia, dysphagia, dysarthria, or muscle weakness with neuroleptic treatment is stronger evidence for an association of drug and disease process because these are not typical side effects of such medications. Our patient developed catatonia, gait disturbance, severe dysarthria and dysphagia, inability to perform tandem gait, and muscle weakness, all of which corresponded temporally to neuroleptic use. Furthermore, withdrawal of the neuroleptics led to improvement in speech and complete resolution of these neurological abnormalities over 24 months without psychiatric decompensation. We could find no other reports of reversible worsening of disease by medication use. At this point it is unclear whether our patient's improvement reflects a resolution of atypical side effects with neuroleptic withdrawal, improvement in the underlying illness secondary to neuroleptic withdrawal, or merely fluctuation in the natural disease progression.
The use of benzodiazepines in the patient we described also raises the possibility of a therapeutic response to benzodiazepines, particularly with respect to his ongoing neurological and psychiatric improvement long after there would have been complete washout of neuroleptics. The efficacy of lorazepam in treating catatonia has been documented.
77,78 Although we know of no other trials of benzodiazepine alone in the treatment of psychiatric symptoms in AGG, our experience with one patient suggests that benzodiazepines deserve consideration as potentially effective therapy in AGG.
There are variable reports of the efficacy of lithium carbonate in treating the psychiatric manifestations of AGG patients. Treatment of postpartum psychosis in an AGG patient with lithium carbonate brought about complete resolution of the delusions within 18 days.
41 Given that this patient had no prior or subsequent psychiatric manifestations, however, it is possible that her psychosis might have resolved over 3 weeks regardless of the addition of lithium. Lithium in combination with either tranylcypromine or carbamazepine has been reported to be effective in several cases.
47,48Use of specific serotonergic reuptake inhibitors has been reported in only 1 patient with AGG; fluoxetine was initiated after electroconvulsive therapy (ECT) to prevent relapse.
47 The long-term benefit of this intervention was not reported.
There are reports suggesting that ECT is efficacious in treating the psychiatric symptoms of AGG.
47,48 An AGG patient with depression had been treated with multiple antipsychotics and antidepressants without benefit.
48 Although a good response to initial ECT was not observed, there was a marked improvement following a further course of ECT. Similarly, another patient with psychosis and depression had only a partial response to multiple medications.
47 Improvement, including resolution of chronic auditory hallucinations, was observed after a course of seven unilateral ECT treatments.
47 Although reports of its use in AGG patients are limited, the available literature suggests that ECT may represent a valuable nonpharmacologic approach to treatment in these patients.