Traumatic brain injury (TBI) affects approximately 200 per 100,000 persons annually in the United States
1 and may produce significant cognitive morbidity among many TBI survivors.
2–8 Attention and memory impairments are generally considered predictable and ubiquitous sequelae of significant TBI,
9–12 and affected TBI patients often describe exacerbation of these impairments in settings of multiple sensory stimuli. Difficulty focusing and sustaining attention in the presence of competing stimuli is sometimes described by clinicians as
distractibility. However, this term as it is often used may encompass at least two forms of attentional impairment. Distractibility is commonly used to describe a rapid shifting of robust selective attention between competing stimuli for brief periods of time (i.e., impaired concentration). Although distractible patients are not able to sustain their attention for long, their ability to select items from a noisy background on which to briefly focus their attention is relatively intact.
13,14By contrast, many patients with attentional impairment after TBI report difficulty in mounting robust selective attention to any stimulus, a problem compounded by the presence of multiple competing stimuli. The problem does not appear to be distractibility per se, but rather an inability to filter out, or “gate,” irrelevant sensory information,
15–17 a phenomenon we describe as
impaired auditory sensory gating.
To make clear the distinction between distractibility and impaired sensory gating: patients with either problem often describe difficulty in conversing with the television on in the background, reading a list in a noisy supermarket, or responding to an interviewer's questions in a room with a noisy fan. When describing this experience in detail, distractible patients will offer a description of each of the distractors in some detail, and they are often able to remember and recount the content of these distractions. For example, while having a conversation in a noisy restaurant, distractible patients may say that they heard part of the conversation with their dinner partner, part of the conversation at the next table, and part of the music being played, and will often be able to relate the details of each part of these events. In other words, although their attention shifts among competing stimuli, they are able to attend to, encode, and later recall the content of the competing stimuli.
Patients with impaired auditory sensory gating also describe their experience in a noisy environment as overstimulating, but they have difficulty recounting the details of the stimuli—they are more apt to say there was “too much going on,” and they are unable to recount in any detail more than the mere presence of the stimuli. Their experience is one of both divided and incomplete attention to all of the stimuli. A metaphor we have found useful for describing the experience of impaired auditory gating is that of listening to a radio receiving two programs on one frequency, resulting in difficulty distinguishing and remembering the information presented by either.
It is plausible that a portion of the attention and memory impairments of persistently symptomatic TBI patients is due to impaired sensory gating,
18–21 since attention is at least in part dependent on such gating mechanisms to filter out irrelevant or excessive sensory inputs.
22 Such sensory gating impairments may be relatively imperceptible to the usual types of testing performed in the practitioner's quiet examination room (e.g., the Mini-Mental State Examination
23), but nonetheless may present the patient with significant difficulties with attention and memory in more usual, and often noisier, environments. However, sensory gating impairments can be identified via electrophysiologic means, including the P50 ratio of the evoked response to paired auditory stimuli (P50 ratio).
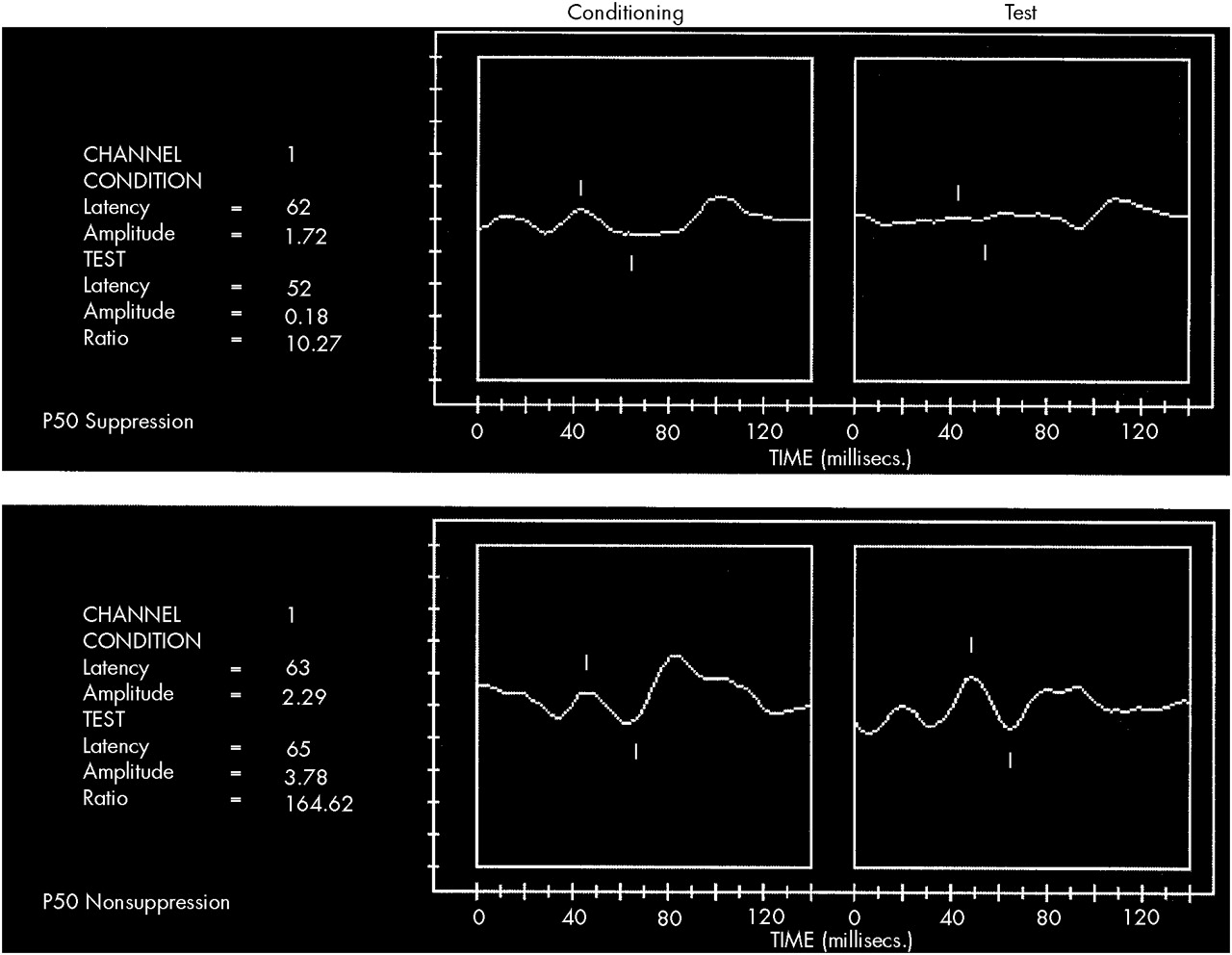
The P50 ratio, an electrophysiologic metric based on comparison of the middle latency auditory cortical evoked potential occurring 50 milliseconds after presentation of paired auditory stimuli presented 500 milliseconds apart, is believed to reflect auditory gating.
22,24,25 Ordinarily when paired auditory stimuli are presented in this fashion, a cortical response is evoked after the first stimulus and an attenuated response is evoked by the second stimulus. A ratio of these responses may be calculated by comparing the amplitude of the P50 evoked by the second or test stimulus with that evoked by the first stimulus. The P50 ratio reflects the action of cholinergically mediated inhibitory neuronal pathways in the hippocampus.
26,27 When these inhibitory pathways are impaired, patients demonstrate abnormally large P50 ratios—that is, the second evoked response is not attenuated (
Figure 1).
Patients with schizophrenia and symptoms consistent with impaired auditory gating demonstrate a significantly increased P50 ratio compared to normal control subjects.
22,25,28,29 These findings have been interpreted as electrophysiologic evidence of persistently disturbed hippocampal cholinergic functioning, an interpretation supported by similar P50 nonsuppression findings in animal models with cholinergic, particularly nicotinic, dysfunction in the hippocampus.
30 In humans, observations of P50 suppression in a previously nonsuppressing subject following nicotine
22,28,31or physostigmine
22 administration also support a cholinergic hypothesis of auditory sensory gating.
20,32,33Auditory sensory gating appears to depend on cholinergic systems, and in particular those acting at low-affinity alpha-7 nicotinic receptors found in the hippocampus.
25,28,32,34 Illness or injury that produces dysfunction of these systems would be expected to produce impaired auditory gating. Because TBI has been shown to damage hippocampal and other neocortical cholinergic systems
35–37 via diffuse axonal injury,
10,38–40 damage to cholinergic neurons,
35–37,41–43 and/or interference with the intracellular mechanisms involved in the synthesis and release of acetylcholine,
44,45 patients with symptoms consistent with impaired auditory gating following TBI would be expected to be nonsuppressors of the P50.
Other electrophysiologic studies have suggested that TBI may produce chronic abnormalities on quantitative electroencephalographic,
5,46–48 evoked potential,
49–52 and event-related potential
53–56 recordings. The P50 ratio may also reflect persistent neurophysiologic disturbance following TBI and might be particularly useful as a marker of both a specific symptom (i.e., impaired auditory sensory gating) and neurophysiological abnormality (i.e., disturbed cholinergic function).
We observed and reported this association between clinical complaints consistent with impaired auditory gating and P50 nonsuppression in three TBI patients seen by the first author.
18 These first three patients varied in level of TBI severity (one each of mild, moderate, and severe) but shared the complaint of impaired auditory sensory gating, prompting speculation that this association might hold for any TBI patient with this symptom. The present study was therefore undertaken to determine whether the phenomenon of P50 nonsuppression could be observed in a larger sample of TBI patients with symptoms of impaired auditory gating. This report describes the first systematic and controlled application of the P50 paradigm to the study of symptomatic TBI patients.
METHODS
Subjects
Subjects were recruited by a combination of direct referral from the first author's neuropsychiatry clinic at our university hospital, referrals from colleagues in the community, and responses to advertisement in a local newspaper. Recruitment efforts solicited the participation of patients who had experienced a TBI at least 1 year ago and subsequently developed symptoms of impaired attention and memory. Potential subjects were initially interviewed by telephone and were excluded if they had any history of psychiatric or neurologic illness prior to TBI, seizures, or alcohol or substance abuse/dependence, or if they were taking medications known to affect measures of auditory gating.
57TBI patients passing the initial telephone interview were subsequently interviewed by the first author. Subjects qualified for participation if they had experienced a definable TBI (nonpenetrating; not requiring neurosurgical intervention; posttraumatic amnesia [PTA] of at least 15 minutes, with or without loss of consciousness) at least 1 year prior to interview and described symptoms consistent with impaired auditory gating. Severity of injury was operationally defined by duration of PTA and was graded as mild (15 min–1 hr), moderate (1–24 hr), or severe (>24 hr)
9 on the basis of patient/family interview and/or record review. This method of assigning TBI severity was chosen because estimation of duration of PTA was more consistently available than Glasgow Coma Scale
58 scores. A Mini-Mental State Examination (MMSE)
23,59 was performed, and subjects were included only if performance was at or above the 25th percentile. The performance of the MMSE was intended to disqualify patients with dementia due to TBI, thereby improving likelihood of historical accuracy generally and with regard to the specific symptoms in question and to better ensure meaningful informed consent and cooperative participation. Severity of persistent disability due to TBI was assessed with the Glasgow Outcome Scale (GOS).
60 Additionally, all subjects were interviewed using DSM-IV
61 criteria to better characterize pre- and post-TBI psychiatric history. Subjects with any definable Axis I disorder prior to their TBI and any active Axis I mood or anxiety disorder at the time of P50 recording were disqualified from participation.
The first 20 subjects (15 women, 5 men) meeting these criteria were enrolled into the study after giving written informed consent, in accordance with the requirements of the Institutional Review Board. Twenty control subjects were selected from a previously acquired normal control population and were included only if they had no history of TBI, had no psychiatric illness known to affect P50 suppression, were on no medications known to affect P50 suppression, and were known to have P50 ratios in the normal range. TBI and control subjects were matched by gender and as closely as possible by age.
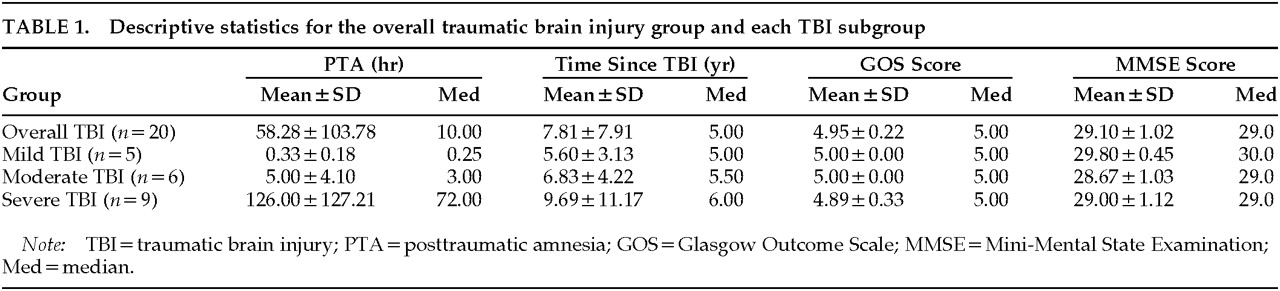
The mean age of the TBI subjects was 42.2 years (SD=11.3), and the mean age of the control subjects was 40.4 years (SD=11.0; difference not significant). Characteristics of the overall TBI group are summarized in
Table 1. There were no differences between the TBI subgroups with respect to time elapsed since initial injury, GOS score, or MMSE score.
Recording Technique
Auditory evoked potentials were assessed according to our previously described methods.
28 Hearing threshold (in decibels) was determined by subjective recognition of the stimuli used in this paradigm at multiple sound pressure levels. Evoked potential recordings were performed with the subject alert, supine, and with eyes fixed on a distant target. EEG activity was recorded on a GRASS Model 78 electroencephalograph with model 7P511 preamplifiers, using a gold disk electrode at the vertex, referenced to one ear. The electro-oculogram (EOG) was simultaneously recorded between the right superior orbit and lateral canthus. The EEG was amplified 20,000 times with bandpass (–50%) between 1 and 300 Hz and was digitized at 100 Hz for acquisition and averaging by using proprietary software on an 80486 IBM-compatible computer. Individual trials were rejected if the EOG or EEG was greater than 50 μV, as such readings indicate movement artifact. The EEG was also monitored for signs of alpha wave activity or other slow waves, and trials demonstrating prominent alpha activity or slowing were rejected.
Auditory stimuli were presented in pairs in a conditioning-testing design with a 0.5-second intrapair interval and a 10-second interstimulus interval. A pulse stimulus approximately 1 ms in duration, amplified with a bandwidth between 20 and 12,000 Hz, was delivered through headphones. The intensity at the subject's ear was set at 30 to 45 dB above hearing threshold. Three sets of average responses were constructed, each based on responses to 16 pairs of stimuli, and were used to create an overall grand average of the conditioning and test P50 responses for each subject.
The P50 wave was identified and measured in each set of averages by using a previously described computer algorithm.
25 The averages were digitally filtered with a 7-point low-pass smoothing routine and a recursive high-pass filter to narrow the bandpass to 10–250 Hz. Each filter was applied twice, in both forward and reverse directions, to increase roll-off and to preserve waveform latency. The algorithm identified the conditioning P50 wave as the most positive peak between 40 and 80 ms after the first stimulus. P50 amplitude was measured relative to the preceding negativity, and a conditioning wave amplitude of at least 0.5 μV was required. The test P50 wave was identified as the most positive peak with a latency from the test stimulus within 10 ms of the latency of the conditioning P50 response, as established in a previous study.
25 The amplitude of the test P50 wave divided by the amplitude of the conditioning P50 wave, expressed as a percentage and referred to here as the P50 ratio, was used as a measure of auditory gating.
Statistical Analyses
Statistica software (Statsoft; Tulsa, OK) was used for all statistical analyses. One-tailed
t-tests of independent samples were used to test for differences in demographic variables, P50 latency, and P50 conditioning and test amplitudes between TBI and control subjects. One-tailed methods were chosen because prior clinical observations of the P50 in patients with TBI suggested that P50 values were likely to vary only in the direction of decreased suppression.
18 Bonferroni-corrected Pearson product-moment correlations were calculated between demographic and evoked potential variables.
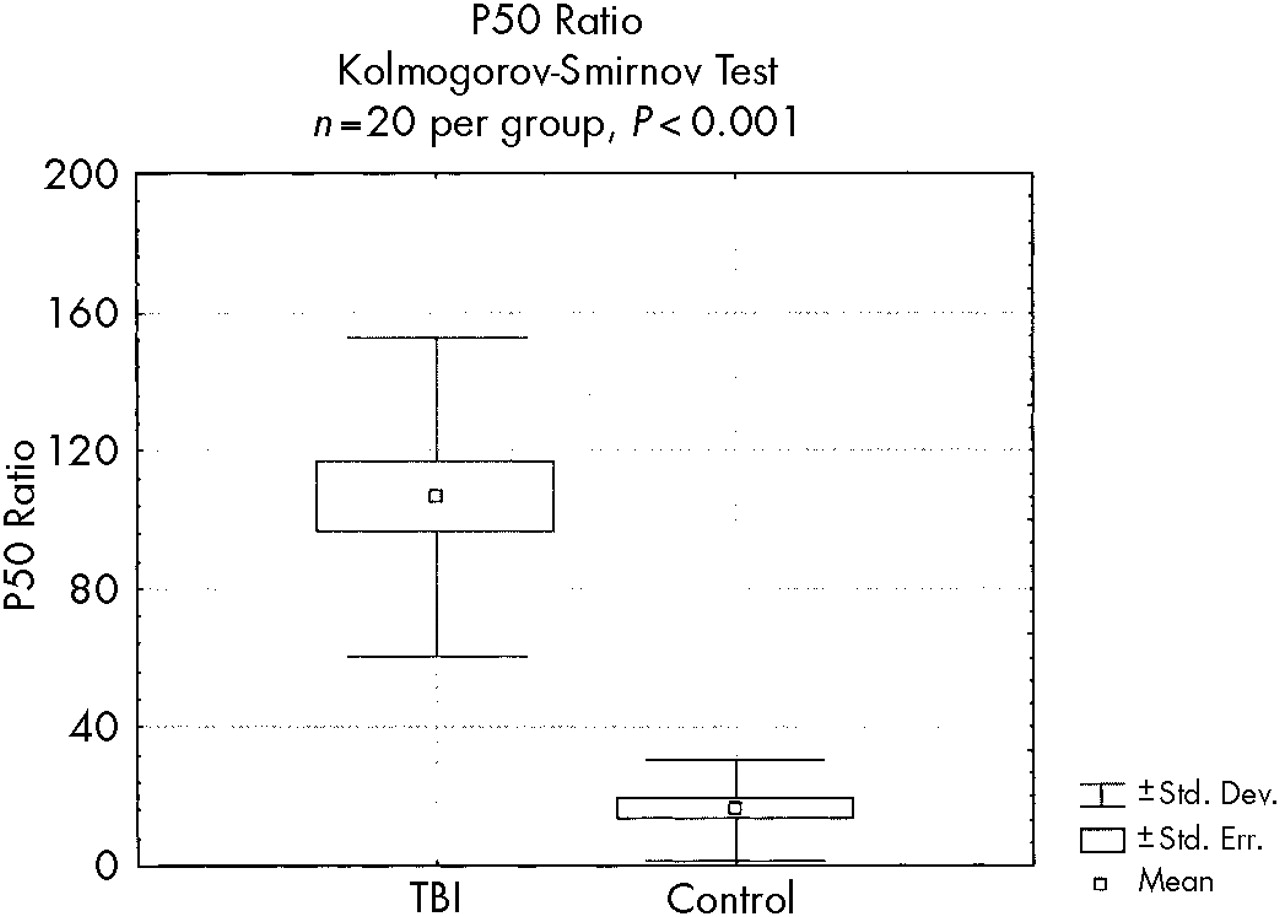
To test for differences in P50 ratios between the TBI and control groups, we used the nonparametric Kolmogorov-Smirnov test. This very conservative method was chosen because it is possible that these groups may represent two different populations (with and without TBI, with and without impaired auditory gating symptoms) and because the general shape of the P50 ratio distributions differed (control subjects clustered between 0 and 20, roughly a truncated normal distribution). Multivariate analysis of variance (MANOVA) was performed to test for differences in demographic variables between each TBI-severity subgroup and control subjects, as well as to test for differences in P50 latency, conditioning and test amplitudes, and P50 ratio. MANOVA was determined to be an acceptable method for this analysis because although the P50 ratio is nonparametrically distributed, this difference derives from alteration most akin to a change in skewness and is therefore unlikely to have a problematic influence on F-value. Post hoc planned comparisons to test for differences in demographic and evoked potential variables between the TBI subgroups were performed by using Tukey's honest significant difference (HSD) test for unequal sample sizes.
RESULTS
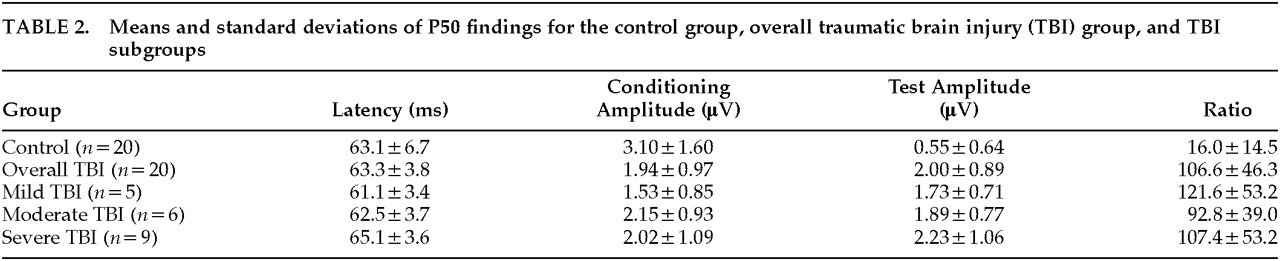
Means and standard deviations for all P50 variables are reported in
Table 2. The P50 ratio was significantly greater in the overall TBI group (Kolmogorov-Smirnov test,
P<0.001;
Figure 2) than in the control group. The conditioning amplitude was significantly smaller (
t=–2.66,
P=0.012) and the test amplitude was significantly larger (
t=5.97,
P<0.001) in the TBI group compared with the control group. Mean latency did not differ significantly between the overall TBI and control groups (
t=0.13,
P=0.89). Bonferroni-corrected correlations revealed no significant relationships between any of the demographic variables and evoked potential variables in the TBI subjects.
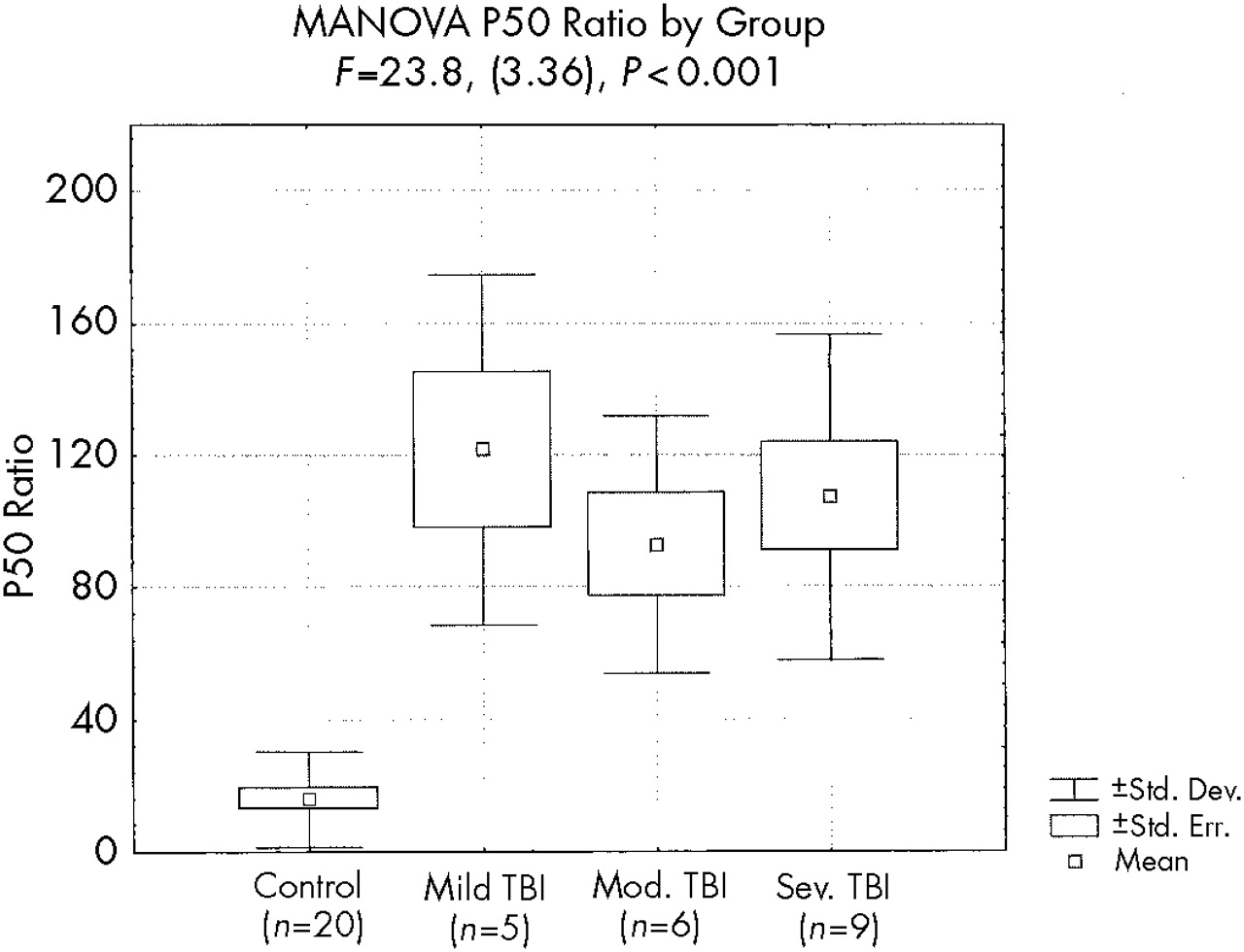
MANOVA demonstrated a significant effect on P50 ratio by group (
F=23.8, df=3,36,
P<0.001;
Figure 3). Post hoc comparisons revealed significant P50 ratio differences between each TBI subgroup and the control group (
P<0.001 for each comparison) but did not reveal significant P50 ratio differences between the TBI subgroups.
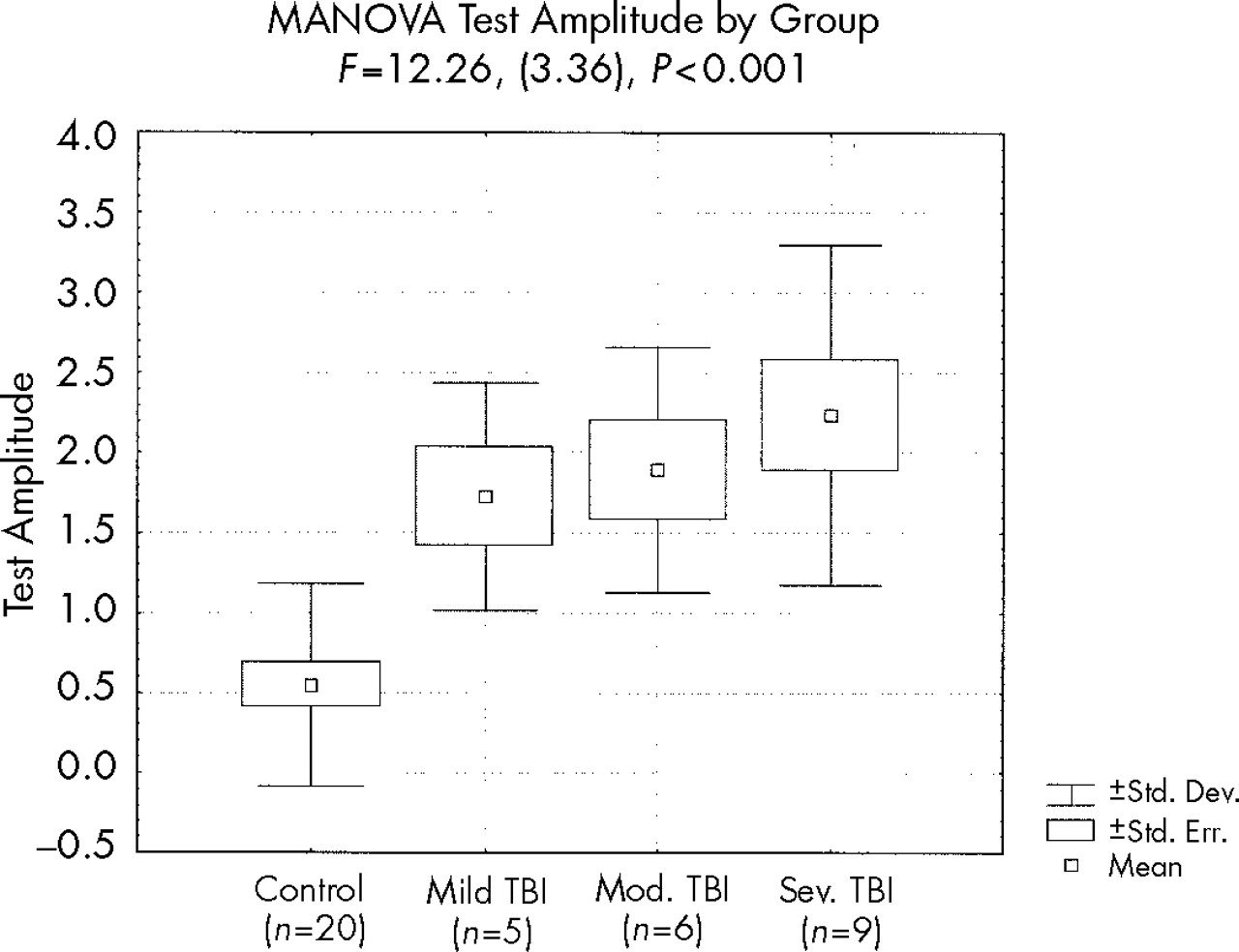
MANOVA demonstrated a significant effect on test amplitude by group (
F=12.3, df=3,36,
P<0.001); post hoc comparisons revealed a trend toward a difference between the mild TBI and control groups (
P<0.09) and significant differences between the moderate TBI and control groups (
P<0.03) and the severe TBI and control groups (
P<0.001), but no differences between the TBI subgroups (
Figure 4).
MANOVA suggested a trend toward an effect on conditioning amplitude by group (F=2.48, df=3,36, P=0.08), with a smaller conditioning amplitude among the TBI groups. Post hoc comparison of conditioning amplitude by group revealed no significant differences between the TBI subgroups and the control group or between the TBI subgroups.
MANOVA revealed no significant effects on demographic variables by group.
DISCUSSION
The present study observed significantly increased P50 ratios among patients who presented with symptoms consistent with impaired auditory gating following TBI. The current subject group was studied because they offered complaints consistent with impaired auditory gating, among many other cognitive complaints, and it was suspected that these complaints might reflect abnormal P50 suppression. This difference in P50 ratio was observed both between the entire TBI group and the control group, as well as between each TBI subgroup (mild, moderate, and severe) and the control group. Given the results of the Kolmogorov-Smirnov test of P50 ratio differences between the overall TBI group and the control group, and the MANOVA and post hoc comparisons of effects on P50 ratio by group, the likelihood of a Type 1 error (false positive) in these results is less than 1 in 1,000. Consequently, P50 nonsuppression among TBI patients with symptoms consistent with impaired auditory gating appears to be a robust finding among patients with mild, moderate, and severe TBI, and it distinguished TBI patients with this symptom from normal control subjects regardless of the level of TBI severity.
The difference in P50 ratio between the TBI and control groups appears to be the result of both a decrease in the conditioning amplitude and an increase in the test amplitude in the TBI groups. The MANOVA analysis of conditioning amplitude by group suggests that the decrease in conditioning amplitude may be a somewhat less robust effect than the increase in test amplitude.
Planned post hoc comparisons of the effects of group on P50 conditioning amplitude, test amplitude, and P50 ratio, using Tukey's HSD test for unequal sample sizes, found no significant differences among the three TBI subgroups based on level of TBI severity. However, although the statistical analysis we used was designed to accommodate the unequal and relatively small sample sizes of the individual TBI subgroups, it is possible that these samples provide inadequate power to find a difference if one exists. Increasing the sample size of each TBI subgroup in subsequent studies is necessary to evaluate more definitively the relationship between level of initial TBI severity and P50 electrophysiology. However, the present data preliminarily suggest that similar levels of P50 nonsuppression may be observed in TBI patients with symptoms of impaired auditory gating despite disparate levels of injury severity and that this phenomenon does not appear to be associated only with severe injuries.
The hippocampus is particularly vulnerable to injury from acceleration/deceleration and rotational forces to which the brain is subjected during TBI.
9,36,38,41,42,62 Because the hippocampus appears to be the structure most strongly related to auditory gating and P50 suppression,
22 and because reports describing P50 nonsuppression in schizophrenia
22,25,63,64 have suggested that hippocampal cholinergic abnormalities substantially contribute to this finding, P50 nonsuppression among TBI patients with symptoms of impaired auditory gating may be consistent with a hypothesis of impaired hippocampal cholinergic function.
18 Although reticulothalamic
51,65–69 and/or reticulofrontal
49,69,70 cholinergic abnormalities may also be contributory, the relative contribution of each system to P50 electrophysiology and to impaired auditory sensory gating has not yet been fully clarified. Nonetheless, the present finding supports the general hypothesis of cholinergic dysfunction in the development of P50 nonsuppression and impaired auditory gating following TBI.
Transient normalization of P50 suppression among patients with schizophrenia has been observed in response to cholinergic augmentation.
22,28,31 Because this deficit is mediated by low-affinity alpha-7 nicotinic receptors, which rapidly depolarize in response to administration of nicotine, the treatment of impaired auditory gating by using cholinergic augmentation strategies with most currently available agents has been problematic.
26,28,31 However, the recent development of centrally selective long-acting acetylcholinesterase inhibitors
71 may make cholinergic augmentation strategies more feasible and potentially beneficial for TBI patients similar to those included in this study. Further, if speculations regarding the potentially deleterious impact of impaired sensory gating on attention, memory, and other aspects of cognition are correct, treatment of impaired sensory gating might be of significant benefit to affected TBI patients.
The present study is limited in its ability to clarify whether P50 nonsuppression specifically indexes impaired auditory gating following TBI or instead indexes significant TBI more generally. Not all TBI patients report symptoms of impaired auditory gating, and therefore not all could be expected to be P50 nonsuppressors. This study examined only patients with symptoms of impaired auditory gating, and thus the prevalence of P50 nonsuppression among TBI patients more generally remains uncertain. It also is not clear whether P50 nonsuppression followed or preceded TBI and what the effect of TBI may be with respect to producing impaired auditory sensory gating if the latter is the case. Additionally, this study cannot establish the strength of the relationship between neuropsychological impairments and these electrophysiologic measures. Because this study was intended only to investigate whether P50 nonsuppression might be observed in TBI patients with complaints consistent with impaired auditory gating, formal neuropsychological testing has not been performed on all of these subjects and is therefore not available to further assist interpretation of these findings. Finally, although the speculation about hippocampal cholinergic dysfunction as the foundation for these symptoms is appealing, at present there are neither structural imaging nor pharmacologic manipulation data to more clearly support such speculations.
Studies designed to address each of these issues are clearly needed to more fully establish the validity of the current finding, the relationship of this finding to neuropsychological and structural neuroimaging variables, and the impact of treatment with cholinergic augmentation on symptoms, neuropsychological performance, and cerebral electrophysiology in TBI patients.