S ince the discovery of the genetic mutation responsible for Huntington’s disease,
1 there have been many attempts to determine whether people who carry the mutation, but who are not yet clinically ill, can be distinguished from those without the mutation. Brain imaging studies have often demonstrated structural and/or functional abnormalities in the striatum in presymptomatic individuals.
2 –
7 In addition, subtle oculomotor and other movement peculiarities and neuropsychiatric changes have been reported in some mutation-positive individuals prior to the onset of clinical signs and symptoms sufficient for diagnosis.
8 –
12 However, the search for presymptomatic changes in cognition among those with the
huntingtin mutation has met with mixed results. Several investigations reported no significant differences in neuropsychological functioning between those with and those without the Huntington’s disease mutation.
13 –
17 Others, however, have found such differences.
8 –
10,
18 Deficits in reaction time, psychomotor and processing speed,
10,
18,
20 –
22 executive functioning,
23 –
25 spatial analysis and constructional praxis,
26 verbal fluency,
23 verbal memory,
26 –
28 and mental arithmetic
9,
10 have all been reported among mutation carriers, albeit inconsistently.
The discrepant findings among studies are due, in large part, to methodological differences. First, studies have varied in the degree of neurologic abnormality tolerated in the “asymptomatic” mutation-positive group.
29 While some researchers enrolled only persons with no, or very minimal, nonspecific neurological signs,
22,
27 others included subjects with “major signs consistent with Huntington’s disease.”
8,
9,
20,
28 In addition, some studies were of young mutation carriers who were almost certainly many years from disease onset, whereas others were of older subjects nearing the age of likely clinical onset. In those studies where proximity to clinical onset was explicitly addressed, time to disease onset (“conversion”) was most often estimated.
10,
15 –
17,
28 The present study is one of the very few reporting data from those who have been followed prospectively from asymptomatic status to diagnosable disease onset.
18,
22 Other factors likely contributing to discrepant research findings include differences in the variety and sensitivity of the specific cognitive tests employed and differences in statistical power to detect group differences (due primarily to sample size).
29We sought to address the limitations of previous work by studying a relatively large group of individuals with the Huntington’s disease mutation who were neurologically healthy at first examinations, who were followed prospectively, and who received the clinical diagnosis of Huntington’s disease at least 2 years after their initial evaluations. We compared the baseline cognitive test performance of these people to that of mutation-positive individuals of equivalent age and education who remained clinically asymptomatic throughout the study period and to at-risk persons who tested negative for the Huntington’s disease mutation. We hypothesized that those who became symptomatic would exhibit greater cognitive impairment at baseline, especially on tests of motor speed, dexterity, and executive control, than those in the other two groups.
Research Participants and Procedure
All participants in the current study underwent presymptomatic genetic testing for Huntington’s disease at the Baltimore Huntington’s Disease Research Center at the Johns Hopkins University School of Medicine and were enrolled in prospective longitudinal studies tracking motor, psychological (cognitive, behavioral and emotional), and brain morphometric changes. The parent study from which these data were drawn was fully reviewed and approved by the Johns Hopkins Medicine Institutional Review Board. All participants gave their informed consent.
At-risk asymptomatic subjects who were first evaluated between February 1986 and January 2007 were eligible for inclusion in these analyses. All were required to have definitive DNA tests for the
huntingtin mutation, complete neuropsychological examinations, and scores ≤10 on the Quantified Neurologic Exam
30 at baseline. The Quantified Neurologic Exam is a standardized assessment of the motor system developed specifically for Huntington’s disease, with higher scores reflecting more movement abnormalities. Scores of barely symptomatic, just-diagnosed patients are typically in the high teens to low 20s.
16,
17 Thus, the single-digit Quantified Neurologic Exam scores of our subjects, and the fact that expert diagnosticians (including AR, RLM, CAR) rated them asymptomatic, ensure that all participants in this study were clinically healthy at entry.
The above criteria were met by 237 people at risk. One hundred thirty-four subjects tested negative for the Huntington’s disease mutation (negative group), while 103 tested positive. Mutation-positive subjects were followed with (in most cases) peri-annual visits. At each visit, the Quantified Neurologic Exam was readministered, as were the battery of neuropsychological tests and several psychiatric and functional rating scales. Of the 103 mutation-positive subjects, only the 70 who were followed for at least 3 years as of June 2007 are included in the analyses reported here; this ensures that they had at least some opportunity to “convert” (i.e., develop diagnosable signs of Huntington’s disease). Of these individuals, 21 in fact converted (converter group) and 49 remained unaffected (nonconverter group) as of June 2007.
RESULTS
The converter, nonconverter, and negative groups were equivalent in age, education, and sex distribution (
Table 1 ). As expected, the groups differed in
CAG repeat length (p<0.001), with the converter and nonconverter groups having more
CAG repeats than the negative group (p<0.001). The difference in
CAG repeat length between converters and nonconverters did not reach statistical significance (p=0.06). Most importantly, all the Quantified Neurologic Exam scores were in the normal range (by design), and neither the difference among the three groups nor the difference between the converters and nonconverters was significant.
For the converter and nonconverter groups, proximity to disease onset was predicted from data available at baseline—namely,
CAG repeat length and parental age of onset.
15 These two groups of mutation carriers did not differ significantly in predicted time to onset, nor did they differ in the length of time they had been followed at the time of these analyses. Subjects in the converter group had actual onset of Huntington’s disease an average of 7.94 years after their baseline visit (SD=4.89, range=1.82–18.28 years); their mean predicted time to onset was 7.12 years (SD=4.61, range=−2.57–15.06 years). These values are not statistically different from each other (p=0.44). The Pearson correlation (
r ) between actual and predicted time to onset was 0.68 (p=0.001). The mean Quantified Neurologic Exam score at the first “symptomatic” assessment for the converter subjects was 15.00 (SD=5.34).
There were no significant differences among the three groups in baseline performance of any of the 10 neuropsychological tests (many tests yielding multiple measures). The largest effect size (η 2 partial =0.029) was on time to complete the Standardized Road-Map Test (F 2,113 =2.40, p=0.094).
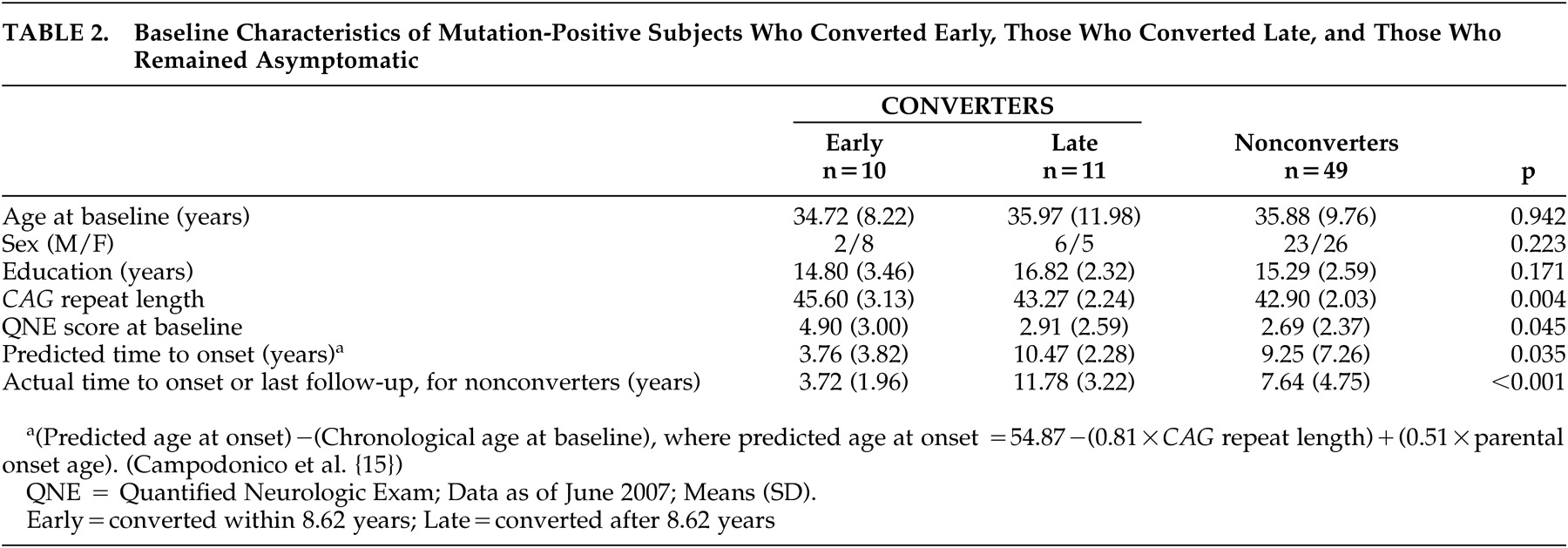
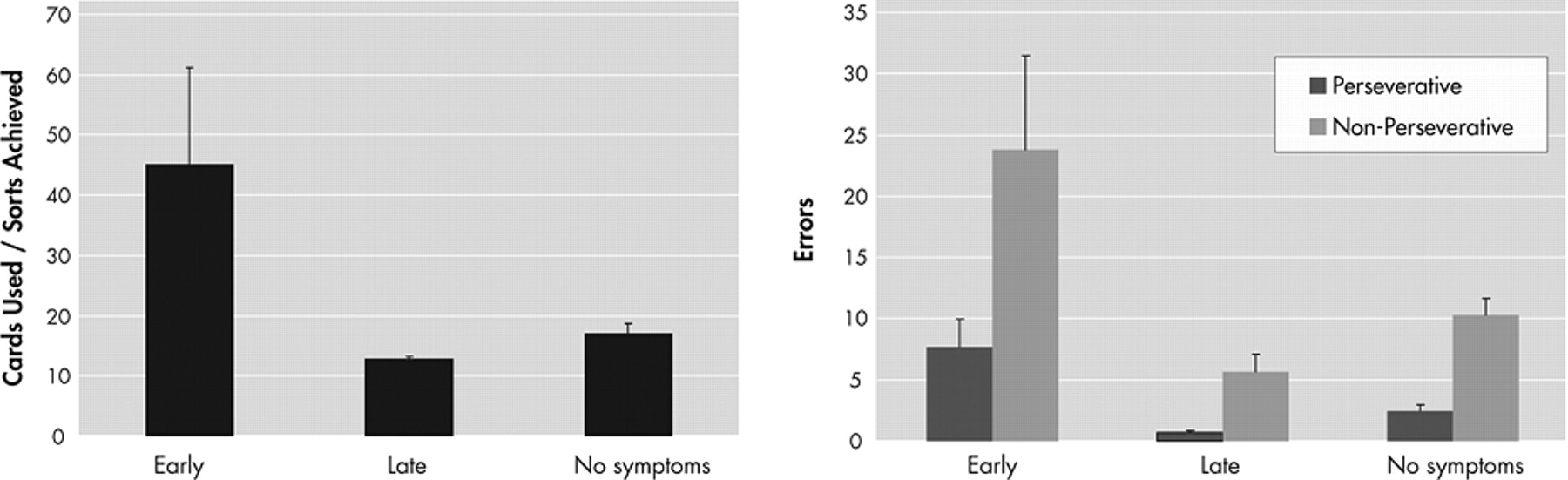
The converter group was divided into those who developed Huntington’s disease soon after their baseline visit (early subgroup) and those who converted later (late subgroup), using the median time to onset of 8.62 years. The early, late, and nonconverter groups did not differ in mean age or education (
Table 2 ). There was a small difference among the groups in mean baseline Quantified Neurologic Exam score, but difference between the early subgroup and late subgroup converters did not reach significance (p=0.07). Examination of the baseline neuropsychological performance of these three groups of participants, all of whom have the Huntington’s disease genetic mutation, revealed significant differences (p<0.01) only on the Wisconsin Card Sorting Test (
Figure 1 ). The early converters differed from both the late converters and the nonconverter groups (who did not differ from each other) in number of sorts achieved (F
2,65 =9.17, η
2 partial =0.220, p<0.001) and in efficiency score (cards used divided by sorts achieved) (F
2,65 =6.53, η
2 partial =0.167, p<0.001). They also differed in the number of both perseverative errors (F
2,65 =6.50, η
2 partial =0.167, p=0.003) and nonperseverative errors (F
2,65 =4.96, η
2 partial =0.132, p=0.01).
DISCUSSION
Most previous studies that have reported cognitive impairment in presymptomatic Huntington’s disease have found it in mutation-positive persons who were
judged to be close to onset.
17 While informative, studies using estimated years to onset provide less definitive evidence of preclinical cognitive changes than investigations of persons who actually convert to Huntington’s disease within a given period. To our knowledge, only two previous studies have followed asymptomatic mutation carriers prospectively until they have become clinically ill. Paulsen et al.
22 analyzed data from the 36-center Huntington’s Study Group and reported that 70 of 260 mutation-positive subjects converted to Huntington’s disease within 2 years of baseline evaluation. These participants performed more poorly on the Symbol Digit Modalities Test and all three trials of the Stroop Color-Word Test (but not on a verbal fluency test) than those who did not convert within 2 years. Notable limitations of this study are the very minimal neurocognitive data available (i.e., three timed tests) and the brief follow-up period. In particular, the fact that all participants converted within 2 years raises the possibility that some persons who were minimally symptomatic were inadvertently enrolled. Snowden et al.
18 compared 51 mutation-positive and 85 mutation-negative subjects on 10 tests, most with multiple outcome measures. Differences emerged only on simple tracing time on the Standardized Road-Map Test (i.e., not making left-right decisions), the color naming trial of the Stroop test, and an alternating hand movement test. These tasks are all described as psychomotor performance tasks that make minimal demands on higher-level cognition, thereby making them particularly sensitive to striatal dysfunction. Twenty-four of the 51 mutation-positives had follow-up assessments, and 15 of them converted to symptomatic at least 5 years after baseline. There were statistical trends for these 15 subjects to differ from the nine who remained unaffected on two variables from the Road-Map Test, time to complete the Wisconsin Card Sorting Test (Nelson modification), and time to complete a picture sequencing task. No mention is made of whether these close-to-onset and far-from-onset groups had equivalent Quantified Neurologic Exam scores at baseline and whether they were followed for the same length of time.
Our study extends the work of Paulsen and Snowden by studying demographically similar samples of asymptomatic mutation-positive and mutation-negative subjects, matched for Quantified Neurologic Exam score, and by comparing presymptomatic mutation-carriers who developed Huntington’s disease at least 2 years later (mean=7.9 years) to well-matched mutation carriers followed the same length of time but who remained unaffected. Under these conditions, we found no evidence of cognitive impairment associated with having the mutation or with converting during the study period. Although we administered many of the same tests as Snowden et al.,
18 we relied on standardized administration. We did not employ the speed modifications Snowden and colleagues made to many tasks, which may have made them more sensitive. On the other hand, Snowden’s mutation-positive and mutation-negative groups differed significantly in baseline Quantified Neurologic Exam score, which may have contributed to the observed group differences in psychomotor performance.
When our converters were stratified into those who developed clinical signs within 8.62 years (the early group, with an average time to conversion of 3.72 years) and those who developed the illness after 8.62 years (the late group, with an average time to conversion of 11.78 years), significant differences were seen on the Wisconsin Card Sorting Test. The early converters achieved fewer sorts, were less efficient (i.e., they required more cards to achieve the sorts they completed), and made more perseverative and nonperseverative errors.
Previous studies from our research group have demonstrated no significant cognitive impairments associated with the
huntingtin mutation when subjects are more than 8 years from predicted disease onset.
15,
16 While no overall group differences were identified between the mutation-positive and mutation-negative groups we described in 2002,
17 a median split of the mutation-positive group on estimated time to onset (8.1 years) yielded significant group differences. Mutation-carriers who were closer to predicted illness onset (mean=4.0 years) performed significantly worse than those further from onset (mean=13.1 years) on the Symbol Digit Modalities Test, WAIS-R Block Design subtest, and all trials of the Stroop test, as well as the Standardized Road-Map Test of Directional Sense and nondominant hand performance on the Grooved Pegboard Test.
In the present study, the converter group was tested an average of 7.94 years before conversion, and the nonconverter group had a predicted time to onset of 9.25 years. Thus, both groups were relatively far from onset at baseline testing. The early converters were tested a mean of 3.72 years before disease onset, and only they displayed selective cognitive deficits on the Wisconsin Card Sorting Test.
Other research groups have identified executive dysfunction in preclinical Huntington’s disease. Lawrence and colleagues
23 reported findings similar to those described here in their study of persons with manifest Huntington’s disease, asymptomatic gene carriers, and mutation-negative comparison subjects. On a modified Tower of London task assessing planning and problem-solving, the gene carriers in this study achieved fewer perfect solutions and required a greater number of attempts to reach solutions than the mutation-negative comparison subjects. In another study, Lawrence et al.
24 reported more errors in attentional set-shifting (an aspect of executive functioning also measured by the Wisconsin Card Sorting Test) in asymptomatic gene carriers compared to mutation-negative subjects. Finally, difficulties with response inhibition have also been documented in preclinical
huntingtin mutation carriers.
25As we see them, the strengths of this study include objectively, operationally defined asymptomatic status of all participants; reliance on actual rather than estimated time to disease onset; extensive follow-up (e.g., some participants followed prospectively for more than 15 years); and relatively comprehensive neuropsychological assessment. Several limitations of the study are acknowledged. First, the number of converters (n=21) remains relatively small. Continued follow-up of our mutation-positive sample should yield additional cases over time. Second, the diagnosis of Huntington’s disease, and the determination of “conversion,” remains a clinical judgment and is subject to error. This is a limitation shared by all studies of the clinical emergence of Huntington’s disease. Finally, we relied heavily on standardized neuropsychological tests. The results of Snowden et al.
18 suggest that research paradigms that stress low-level, automatic, cognitive and motor routines may be more sensitive than clinical tests on the earliest brain changes in Huntington’s disease.
Our finding of a highly selective presymptomatic cognitive change among those “incubating” Huntington’s disease parallels recent reports of changes in brain morphology and functioning prior to clinical disease onset. Research from our group has revealed progressive decreases in the volume of the caudate nucleus on structural MRI starting 9–11 years prior to clinical onset.
2 Other structural changes have been reported in cerebral white matter
40 –
41 and selected cortical regions.
3 Studies using PET imaging have revealed decreased activation at rest in the frontal and temporal cortex
40 as well as decreased D
2 receptor binding in the striatum.
42 Functional imaging studies with cognitive activation have found reduced activation in subcortical regions (caudate and thalamus),
4 as well as increased activation in medial prefrontal cortex
4 or decreased activation in the anterior cingulate cortex,
5 probably depending on the task. Such changes in the structure and function of fronto-striatal circuitry are consistent with the executive dysfunction demonstrated in our asymptomatic gene carriers.
In conclusion, this long-term prospective study of neurologically healthy persons with the Huntington’s disease mutation found that those who develop symptoms an average of 7.9 years after baseline assessment displayed no cognitive impairments. However, those who converted soon after baseline (within 8.6 years), as a group, performed more poorly on a problem-solving test requiring mental flexibility and concept formation than those who developed the disease later. This impairment is by no means sufficiently robust to be used in counseling individual cases, but it does support prior observations that the huntingtin mutation has phenotypic expression prior to frank disease onset.