Neuropsychiatric conditions abound in these neurodegenerative diseases, and psychotropic drugs are routinely applied in treating them. Aggression, agitation, anxiety, apathy, depression, disinhibition, and psychosis are among the many behavioral complications in neurodegenerative diseases, are highly prevalent, and add tremendously to patient distress and caregiver burden.
1 –
29 Psychotropics are often prescribed to control neuropsychiatric behavioral disturbances in these diseases.
2,
3,
8,
9,
11 –
16,
18,
19,
23,
29 –
38 Thus, it is fortuitous that drugs that provide clinical benefit in neurodegenerative diseases may also afford the neuroprotective capacity to deter the progression of underlying pathobiologies.
In Part I of this report, we reviewed preclinical effects of psychotropics on selected pathophysiological processes operant in neurodegenerative disease. Here, we review the broader diversity of neuroprotective actions and the potential effects of selected psychotropics representing pharmacological classes of drugs with promising neuroprotective potential. These psychotropics are selected to represent pharmacodynamic drug classes with multiple neuroprotective actions. We will overview these neuroprotective actions and provide some examples of the complexity of their interactions.
A Diversity of Neuroprotective Actions
A wide array of mechanisms provide potential opportunities for neuroprotective treatment in the neurodegenerative diseases. Briefly, the enzyme
glycogen synthase kinase-3 (GSK-3) promotes
alpha-synuclein (αSyn) expression,
39 beta-amyloid (Aβ) production,
40 and
tau phosphorylation.
39,
41 The neurodegenerative diseases are linked to pathological protein accumulation, for example, αSyn, particularly in Parkinson’s disease,
42 Parkinson’s disease dementia,
43 and dementia with Lewy bodies,
43 and Aβ and hyperphosphorylated tau, especially in Alzheimer’s disease.
42,
44,
45 The newly emergent transactive response RNA/DNA binding protein
TDP-43 affects RNA splicing
46 and is the dominant component of ubiquinated inclusions present in the TDP-43 proteinopathies, including frontotemporal lobar degeneration with ubiquitin immunoreactive inclusions (FTLD-U) and amyotrophic lateral sclerosis,
47 although psychotropic effects on TDP-43 remain to be discovered. It is these pathogenic protein inclusions that form the pathological stigmata of the neurodegenerative diseases, namely, Lewy bodies (αSyn) in Parkinson’s disease and amyloid plaques (Aβ) and neurofibrillary tangles (hyperphosphorylated tau) in Alzheimer’s disease.
Interestingly, treatments lowering plasma Aβ are thought to reduce the risk of developing Alzheimer’s disease,
48 –
50 and it is likely that modulation of plasma αSyn, Aβ, and tau concentrations can similarly lower the risk of developing other neurodegenerative diseases. Consistent with this concept, plasma αSyn oligomers are increased in Parkinson’s disease
51,
52 and in multiple system atrophy.
52 Just as the plasma Aβ 42/40 forms ratio falls at the onset of Alzheimer’s disease,
53,
54 plasma αSyn concentrations also appear to fall with the advent of Parkinson’s disease,
55 presumably attributable to deposition within the CNS. Consequently, reductions in plasma pathogenic proteins may, in the future, translate to reduced brain neuropathology in a number of neurodegenerative diseases.
Pathogenic proteins can lead to
proteasomal 56 –
58 and
mitochondrial dysfunction,
59,
60 increase
intracellular calcium 61 and
free radicals,
62 and compromise
axonal transport and
cytoskeletal integrity,
63 leading to
apoptosis .
The proteasome degrades damaged, toxic, excessive, and unwanted proteins from cells.
56 Undesirable proteins are tagged with a poly-ubiquitin chain by the E1-E3 ubiquitin conjugase-ligase system.
56 Poly-ubiquitylated proteins then enter the proteasome for degradation. The 26S proteasome consists of a 20S proteolytic core capped by 19S caps.
56 Unwanted proteins enter the caps, ubiquitin chains are cleaved, and the protein is unfolded and fed into the proteolytic core for cleavage into peptide fragments. It is interesting to note that the ubiquitinopathic frontotemporal dementia FTDU-17 is associated with tau-negative, ubiquitin-positive inclusions, and that mutant PARK 2 autosomal recessive juvenile Parkinson’s disease
parkin has E3 ubiquitin ligase activity (part of the E1-E3 ubiquitin conjugase-ligase system). Oxidative stress impairs both ubiquitylation and proteasomal function, and proteasomal dysfunction engenders additional oxidative stress and free radical formation.
64 Proteasomal failure further leads to impaired degradation of pathogenic proteins, apoptotic mediators (e.g., caspases and bcl-2 proteins),
65 and regulators of apoptotic transcription factors (e.g., p53, NF-kappaB, HIF-1 alpha),
65 reduced axonal viability and synaptic integrity,
66 and mitochondrial dysfunction.
60Mitochondrial dysfunction leads to the generation of
reactive oxygen species and consequent
mitochondrial depolarization and
mitochondrial permeability transition pore development with the release of
free radicals and
cytochrome c into the cytoplasm, which in turn trigger the apoptotic cascade.
67 Mitochondrial respiratory dysfunction, including complex I
68 and complex II
69,
70 deficiencies and the complex I inhibitors rotenone
71 and tumor necrotic factor,
72 each leads to apoptosis. Additionally, free radicals including reactive oxygen species and reactive nitrogen species result in lipid peroxidation that advances the course of neurodegenerative disease.
73,
74 In Parkinson’s disease, free radicals are linked to dopaminergic neuronal loss.
75 Reactive species include peroxide radicals, especially linked to dopamine oxidation and superoxide dismutase, and nitric oxide, especially related to inducible nitric oxide synthase, constituting major mechanisms of interest in neurodegenerative diseases.
73,
76 Nitric oxide promotes the generation of free radicals,
77 which in turn advance the progression of neurodegenerative diseases.
78There are several apoptotic pathways that play key roles in neurodegenerative diseases, including mitochondrial-, death receptor-, and p53-mediated apoptotic pathways.
79,
80 The mitochondrial pathway involves cytochrome c-related activation of caspase-9, which activates caspase-3
81 and the apoptotic cascade.
82 This mitochondrial apoptotic pathway plays a principal role in neurodegenerative diseases.
83 The death receptor, or Fas pathway, involves FADD and caspase-8 activation, whereas the p53 pathway involves glyceraldehyde-3-phosphate dehydrogenase and Bax, with caspase-8 and Bax initiating the apoptotic cascade.
79,
80In addition,
glutamate excitotoxicity is a leading theory of neurodegeneration in Alzheimer’s disease,
84 Parkinson’s disease,
85,
86 Huntington’s disease,
87,
88 and amyotrophic lateral sclerosis
89,
90 and is a basis for the potential neuroprotective effects of memantine
84,
91 and riluzole.
92 Glutamate mediates apoptosis through mechanisms that include glutamatergic
N -methyl-D-aspartate (NMDA) receptors,
93,
94 calcium influx,
95 and free radicals.
93,
95,
96 Inhibition of GSK-3β protects against glutamate-induced, caspase-3-mediated apoptosis.
97Inflammatory processes are also evident in the neurodegenerative diseases. Aβ neuritic plaques and proinflammatory cytokines in Alzheimer’s disease lead to
microglial activation and
inflammation .
98 Activated microglia, reactive astrogliosis, and lymphocytic infiltration are also apparent in the substantia nigra in Parkinson’s disease as a later concomitant of neurodegeneration,
99,
100 possibly related to αSyn and oxidative stress.
64,
100 Activated microglia then produce cytokines, reactive oxygen and nitrogen species, and eicosanoids that propagate further neurodegeneration.
101 It is interesting to note that sleep loss may promote inflammation.
102 Sleep loss is often present in neurodegenerative diseases, especially related to fragmentary daytime napping, depression, psychosis, or insomnia.
These neurodegenerative mechanisms occur in the absence of neurodegenerative diseases and are balanced by neuroregenerative processes, including neuritogenesis and the influence of neurotrophic factors.
Cytoskeletal derangements occur in neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease related to changes in tau and tubulin, and there is evidence that certain drugs and hormones can affect this and stimulate
neuritogenesis .
103 Neuritogenesis is key to neuronal and synaptic recovery and regeneration after injury and is a treatment target in neurodegenerative diseases.
104 Neurotrophic factors include brain-derived neurotrophic factors (BDNF) and glial-derived neurotrophic factors (GDNF). BDNF
105,
106 and GDNF
105 –
110 consitute important treatment targets in Alzheimer’s disease,
106 Parkinson’s disease,
106,
108 –
110 Huntington’s disease,
105 amyotrophic lateral sclerosis,
107 and spinal muscular atrophy,
107 with GDNF showing more potential than BDNF in most models.
105,
107,
109 Persephin and other GDNFs have promoted neuronal survival and neuritogenesis in a midbrain dopamine neuron model of Parkinson’s disease.
111Complex Interactions Between Neurodegenerative Mechanisms
The number of neuroprotective mechanisms is complex. The interaction of various mechanisms with each other is also complex. For example, proteasomal inhibition increases the accumulation of pathogenic proteins, impairs mitochondrial function, and triggers apoptosis. Further complexity of interaction is apparent in considering specific mechanisms, for example, pathogenic proteins.
It has been found that Aβ and tau each facilitate αSyn aggregation in Parkinson’s disease
112 –
114 and that αSyn is linked to Alzheimer’s disease
115 and facilitates tau aggregation in Alzheimer’s disease.
112 αSyn and tau each independently initiate amyloid formation.
116 Moreover, GSK-3 promotes Aβ formation by phosphorylating amyloid precursor protein
40 and also promotes tau hyperphosphorylation to pathogenic tau
41 in Alzheimer’s disease. In Parkinson’s disease, GSK-3 alleles are associated with Parkinson’s disease risk.
113,
114 Furthermore, GSK-3β inhibitors may reduce αSyn
39 and αSyn upregulates GSK-3β,
117 suggesting that GSK-3β and αSyn mutually upregulate each other in Parkinson’s disease models. These pathological proteins interact at various levels in the pathological chain of events that involves protein processing at the proteasome, mitochondrial destabilization, free radical generation, apoptotic pathway activation, cell death, and neuroinflammation.
GSK-3, αSyn, tau, and Aβ each inhibit the proteasome,
56 impair mitochondrial function,
59,
60 generate free radicals,
62 and result in apoptosis.
42,
118 These processes are compounded by their induction of microglial activation
98 –
100 and neuroinflamation.
101 Mutual upregulation of αSyn and GSK-3β in Parkinson’s disease can lead to apoptosis by GSK-3β-induced bcl-2 downregulation,
117 and by synphilin-1 phosphorylation
119 that induces endoplasmic stress and proteasomal dysfunction
119 in Parkinson’s disease. Also, in Parkinson’s disease models, GSK-3 further provides pathways through which rotenone,
120 6-hydroxydopamine,
121 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP),
120,
122 and
l -dopa
123,
124 mediate apoptosis. Finally, apoptosis is also indirectly triggered by tau and Aβ
118 in both Alzheimer’s disease and Parkinson’s disease
42 by inhibiting proteasomal
56 and mitochondrial
60 function and inducing free radical damage,
62 microglial activation,
98 –
100 and inflammation.
101These relationships of pathogenic proteins to specific neurodegenerative mechanisms are just one example of the complexity of interaction between various components of the neurodegenerative process. Thus, neuroprotective strategies that address individual neurodegenerative mechanisms can potentially enhance their neuroprotective effects through these complex interactions.
Neuroprotection Targets
It is apparent from the above discussion that a number of independent mechanisms produce neurodegeneration. Consequently, a wide variety of neuroprotective targets exist and these include GSK-3, glutamate, tau hyperphosphorylation and aggregation, Aβ production and aggregation, αSyn production and aggregation, other protein (e.g., TDP-43) production and aggregation, proteasomal function, free radical generation (reactive oxygen, nitrogen, and other species), oxidative stress, antioxidant enzymes, mitochondrial dysfunction (including complex I, complex II, other respiratory chain components, and ATP production), apoptosis, neuroinflammation, cytoskeletal integrity, trophic factors (e.g., BDNF, GDNF), and neuritogenesis.
Scope of the Present Report
The findings in Part I of this report indicate possible neuroprotective applications for psychotropics in neurodegenerative diseases. Here, we integrate those findings and consider some neuroprotective candidate agents that are used to treat neuropsychiatric disturbances in the clinical arena and, based on their pharmacological class membership, may have salutary effects at various levels of the neurodegenerative pathophysiological sequence. In Part I (Winter 2010 issue, available at http://neuro.psychiatryonline.org/), we considered candidate neuroprotective agents based upon research findings for first-line psychotropics in regard to only certain neuroprotective mechanisms. In contrast, here we consider the broader diversity of neuroprotective mechanisms for representative drugs that exert pharmacodynamic actions consistent with those of promising agents discussed in Part I. We further consider the wider spectrum of neuroprotective attributes for the selected psychotropics, building upon the findings in Part I of this report.
An important caveat to this synthesis is that the neuroprotective actions of these psychotropics may not necessarily be mediated by their established pharmacodynamic mechanisms. While there is early evidence that at least some of their actions are in fact mediated by these pharmacodynamic actions, this literature is in its infancy. Further, although representative drugs are put forth, supporting evidence is drawn from drugs within their pharmacological class possessing identical primary pharmacodynamic actions. Nevertheless, there are common neuroprotective effects within given psychotropic classes (see Part I of this report), and some of these have already been linked to specific pharmacodynamic mechanisms (e.g., serotonin, melatonin receptors, etc.).
The drugs considered here appear to have been neglected and subjected to little research thus far, as is evident in Part I of this report. They are selected based on evidence in Part I of this report supporting the neuroprotective potential of psychotropic drugs possessing their mechanisms of action. We consider representative drugs of pharmacological classes that have symptomatic utility in neurodegenerative diseases and should now be subjected to preclinical investigations according to the criteria in
Table 1 . Based on these criteria, research using delayed-start (staggered start) and randomized-withdrawal designs
125 can be undertaken to determine their disease-modifying neuroprotective utility. Examples of wakefulness promoting, anxiolytic, antidepressant, antipsychotic, and hypnotic agents are provided.
METHODS
The method is specified in Part I of this report but, briefly, it relied on the Part I search strategy extended by bibliographic review and other sources including literature searches of peer reviewed published articles for the drugs specified and for neuroprotective actions ranging beyond those focused on in Part I.
Modafinil
Modafinil is a drug with symptomatic potential in neurodegenerative diseases that binds to the dopamine transporter
126 –
128 and increases synaptic DA.
127 It may act as a direct dopamine D2 receptor agonist
129 and may secondarily stimulate noradrenergic activity.
130 –
132 Dopaminergic and noradrenergic deficits occur in Alzheimer’s disease and Parkinson’s disease, and these modafinil actions could improve daytime somnolence, sundowning, cognition, and neuropsychiatric disorders in dementia. Modafinil has effectively treated excessive daytime sleepiness in Parkinson’s disease
133 and amyotrophic lateral sclerosis.
134 Performance on the clock drawing task, which integrates executive, visuospatial, and other cognitive functions that are impaired in dementia and is commonly used to screen for dementia, improved in healthy middle-aged subjects given modafinil.
135 Modafinil has also been demonstrated to increase hippocampal glutamate release.
136 Glutamate is critical for proper memory function through the process of long-term potentiation.
137,
138 Increasing glutamate release may lead to improved memory function in disorders where the hippocampus is degenerating. In various disorders, modafinil has improved executive function,
139 –
144 verbal fluency,
145 attention,
139,
144,
146 –
150 memory,
140,
141,
144 apathy,
151,
152 fatigue,
153 depression,
154 –
157 and response inhibition.
139 –
141,
144 Improvements in cognition and response inhibition may further improve impulsivity, disinhibition, and aggression. Moreover, modafinil has been suggested to potentially improve Parkinson’s disease symptoms related to enhanced glutamatergic striatal stimulation and reduced striatopallidal GABA release.
158Modafinil appears to simultaneously enhance glutamatergic symptomatic function while blocking its neurotoxic effects,
159 consistent with a neuroprotective action potentially applicable in a number of neurodegenerative diseases, including Alzheimer’s disease, Huntington’s disease, and amyotrophic lateral sclerosis. Modafinil also dose-dependently
160 blocks MPTP neurotoxicity in the marmoset monkey Parkinson’s disease model
160,
161 through antioxidant effects and the modulation of nigrostriatal monoamines.
162 This same effect has been observed in nigral neuronal and glial cells in black mice.
163 Thus, modafinil neuroprotective properties against glutamate toxicity and nigral degeneration may translate to clinical neuroprotection in patients with neurodegenerative diseases. The potential neuroprotective actions of modafinil are summarized in
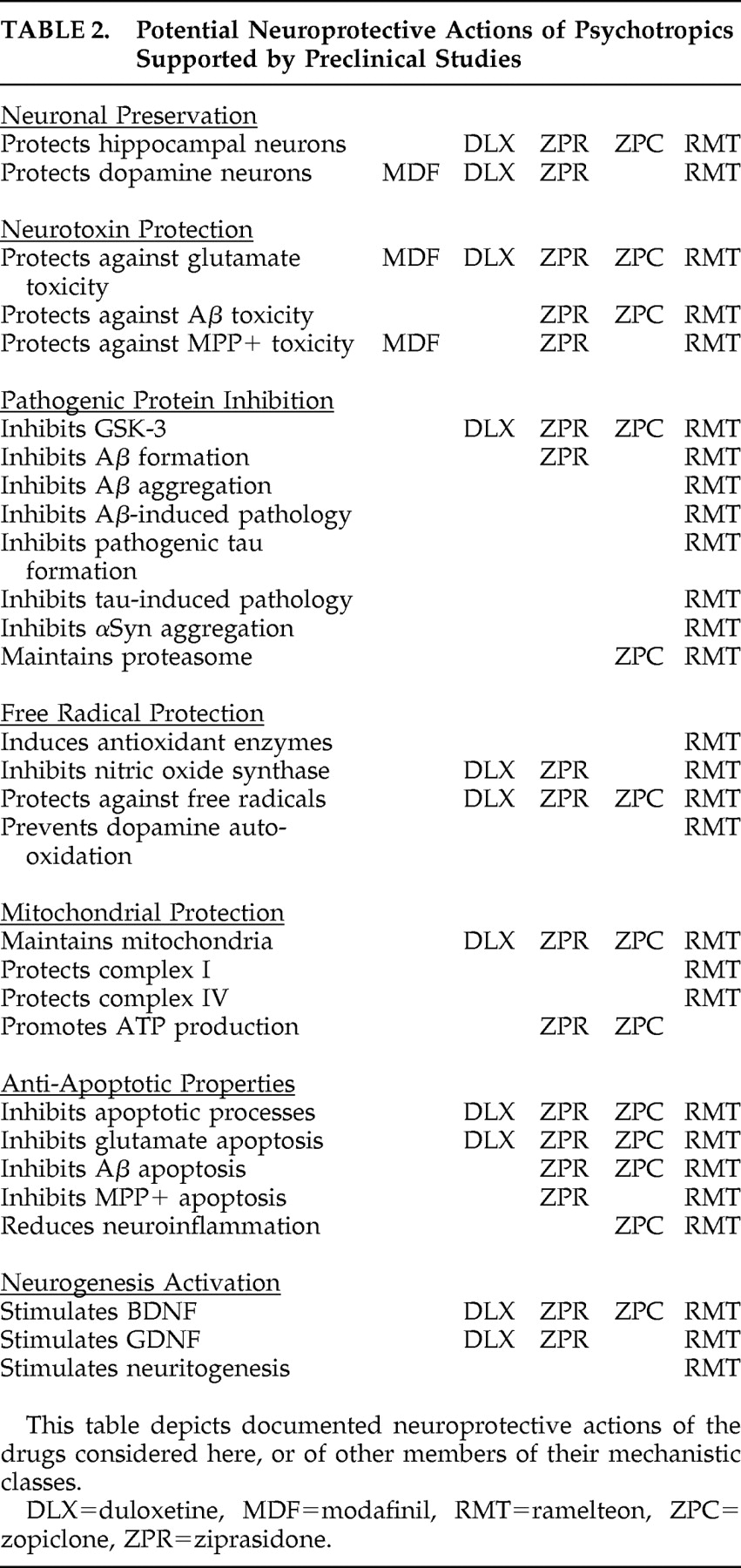
Table 2 .
Duloxetine
The antidepressant duloxetine is a dual serotonin and norepinephrine reuptake inhibitor (SNRI).
164 Duloxetine has improved several measures of depression and cognition in a double-blind, placebo-controlled trial in geriatric patients.
165 Experience with selective serotonin reuptake inhibitors (SSRIs) and norepinephrine reuptake inhibiting antidepressants, together with neurotransmitter correlate findings for certain neuropsychiatric disorders, suggests that duloxetine has the potential to improve depression,
165 –
175 anxiety,
15,
16,
166,
172,
176 hallucinations,
177 –
181 delusions,
177,
181,
182 sleep,
183,
184 agitation,
166,
181,
182,
185,
186 aggression,
181,
182,
186 irritability,
166,
176,
181,
182,
186,
187 disinhibition,
188,
189 excitement,
176,
182 wandering,
182 overall cognition,
165,
190 –
193 attention,
194 –
196 executive functioning,
197 and memory
165 across neurodegenerative diseases. Apathy and urinary incontinence may also respond to duloxetine. Apathy and other dopamine-related symptoms can respond to duloxetine because norepinephrine reuptake inhibitors block frontal dopamine reuptake.
198 Indeed, apathy responds to agents that increase frontal dopamine.
199,
200 Similarly, in Parkinson’s disease, neurotransmitter correlates and agents that affect neurotransmitters further evidence the potential for improvements in Parkinson’s disease visuospatial processing
201 as well as bradyphrenia, akinesia, postural instability, freezing, dyskinesia, and Parkinson’s disease stage.
202 –
206 Urinary incontinence in dementia usually results from either sphincteric or detrusor muscle dysfunctions. Duloxetine is thought to work centrally at Onuf’s nucleus in the sacral spinal cord to improve sphincter tone mediated through 5HT2 and alpha-1 receptor effects
207 during urine storage but not during voiding, thereby significantly improving stress urinary incontinence.
208,
209 In addition, a recent placebo-controlled study in women with overactive bladders due to documented detrusor instability revealed improved detrusor stability with reduced episodes of incontinence and voiding. This presumably is related to raising the sensory threshold for triggering micturition mediated by the 5HT1a receptor at the primary afferent neurons in the superficial dorsal horn of the sacral spinal cord.
210Duloxetine and agents with related modes of action together have demonstrated neuroprotective properties in neurodegenerative models relevant to neurodegenerative diseases, such as reducing pathogenic tau and Aβ, proteins that also facilitate and synergize αSyn pathogenic oligomeric fibrillation and promote proteinopathic inclusions in Alzheimer’s disease and Parkinson’s disease. Other properties include inhibition of GSK-3β (thereby reducing αS, tau, and Aβ formation) and reductions in free radical production, nitric oxide synthase activity, mitochondrial permeability transition pore development, apoptosis (especially 6-hydroxydopamine- and glutamate-induced apoptosis in Parkinson’s disease and Alzheimer’s disease models), microglial activation, and inflammation. These drugs also upregulate BDNF and GDNF. We will sequentially summarize the evidence for SSRIs, selective norepinephrine reuptake inhibitors (NERIs), and SNRIs.
SSRIs reduce Aβ and tau concentrations and inhibit GSK-3β and apoptosis, including in the hippocampal formation. The SSRI paroxetine preserved cognitive performance and reduced hippocampal Aβ and tau levels in transgenic mice.
211 Additionally, antidepressants have effects on GSK-3, affecting neuronal pathological protein concentrations. The SSRI fluoxetine
212 and the SNRI imipramine
213 have each inhibited GSK-3. SSRI-induced GSK-3β inhibition appears to be mediated by 5HT1a receptor stimulation, which inhibits GSK-3β through serine 9 phosphorylation.
214 SSRIs improve cell proliferation in several models and inhibit several types of apoptosis in a variety of cell lines. Fluoxetine improved contextual memory and cell proliferation in Aβ transgenic mice.
215 Fluoxetine,
216 –
220 clomipramine,
220 paroxetine,
218 and citalopram
218 have inhibited apoptosis in neoplastic models. Of particular relevance in Alzheimer’s disease, fluoxetine enhanced cell proliferation and prevented dentate gyrus apoptosis in a rat model.
221Selective NERIs block free radical generation including intramitochondrial radicals, mitochondrial permeability transition pore development, and several types of apoptosis in various cell lines. The selective NERI desipramine
222,
223 has inhibited HDAC inhibitor/perifosine-induced
224 and tumor necrotic factor
a -induced
225,
226 reactive oxygen species production, glutamate-induced mitochondrial permeability transition pore opening in Huntington’s disease transgenic mice,
227 and apoptosis in a wide variety of models.
224 –
226,
228 –
234 These apoptotic models include glutamate in transgenic Huntington’s disease mice
227 and 6-OHDA in neuronal PC12 cells relevant to Parkinson’s disease.
235,
236SNRIs like duloxetine inhibit GSK-3β, nitric oxide synthase, mitochondrial permeability transition pore opening, and several types of apoptosis in various cell lines. Duloxetine inhibits nitric oxide synthase
237 and, hence, free radical generation. The SNRI nortriptyline blocked glutamate-induced mitochondrial permeability transition pore opening and apoptosis in a transgenic mouse Huntington’s disease model.
227 As noted above, glutamate is also key to Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Imipramine inhibited tumor necrotic factor
a -induced apoptosis
238 and cisplatin-induced clustering of CD95 apoptotic death receptors.
239All three types of antidepressants reduce microglial activation and the expression of interleukin-6 and nitric oxide,
240 and several antidepressants including duloxetine upregulate the neurotrophic factors BDNF and GDNF to produce ex vivo and in vivo neuroprotection. BDNF is reduced in the frontal
241 and temporal
242 cortex and hippocampus
242 in Alzheimer’s disease. Chronic administration of duloxetine upregulates BDNF mRNA in frontal cortical synaptosomes, providing the potential to maintain or improve frontal cognition.
241,
243 In Parkinson’s disease, BDNF is colocalized with dopamine neurons in the substantia nigra where it can function neuroprotectively, and it may have a beneficial neuromodulatory action as well.
244 Plasma BDNF levels were significantly lower in depressed patients than in healthy control subjects and normalized after 1 month of antidepressant treatment.
245 The same phenomenon has been demonstrated for serum GDNF,
246 a target in most neurodegenerative diseases. Antidepressant-induced increases in GDNF have been shown to be neuroprotective in both ex vivo
247 and in vivo
248 models and are mediated through a monoamine-independent mechanism.
110,
249 Each of these effects is particularly relevant to Parkinson’s disease pathogenesis
64 and has the potential to protect against neurodegeneration in Parkinson’s disease and other neurodegenerative diseases. In principle, duloxetine offers the potential to combine the symptomatic and neuroprotective properties of the different antidepressants into an effective neurodegenerative disease treatment. The potential neuroprotective actions of duloxetine are summarized in
Table 2 .
Ziprasidone
Ziprasidone is an atypical antipsychotic with D2 antagonist, 5HT2 inverse agonist, and 5HT1a agonist properties, as well as SNRI activity.
250,
251 Ziprasidone modes of action thereby confer antipsychotic, antidepressant, and anxiolytic properties translating to a broad therapeutic potential in neurodegenerative diseases.
Aside from antipsychotic effects on hallucinations and delusions,
252 other ziprasidone benefits include effects on agitated behavior,
253 –
256 anxiety,
257 depression,
250,
251,
258,
259 sleep,
260 and cognition.
251,
261 Although results from controlled studies in dementia are lacking, in three frail elderly patients with dementia, ziprasidone was effective in improving psychosis, agitation, depression, and cognition in each case.
262 Additionally, improvement in negative symptoms
263,
264 and cognition
266 –
268 in schizophrenia trials suggests that apathy and cognition (particularly executive function, working memory, episodic memory, attention/vigilance, and psychomotor speed) may improve in patients with dementia. Moreover, MMSE scores
269 and working memory
270 are sensitive to serotonin availability. Furthermore, as referenced in the duloxetine section, the dual SNRI action of ziprasidone suggests specific additional nonantipsychotic-mediated therapeutic effects on a wide variety of symptoms. These include hallucinations, delusions, depression, anxiety, apathy, sleep, agitation, aggression, irritability, disinhibition, excitement, wandering, incontinence, overall cognition, attention, executive function, memory, and visuospatial function. In Parkinson’s disease, at doses low enough to invoke ziprasidone actions other than D2 blockade, it is possible that beneficial effects may be seen on bradyphrenia, motor symptoms including akinesia, postural instability, freezing, stage, disability, dyskinesia, and on-off syndrome. In contrast to most other atypical antipsychotics, early clinical evidence suggests minimal motor impairment in Parkinson’s disease with ziprasidone.
255,
271Ziprasidone and agents with related modes of action together demonstrate neuroprotective properties in neurodegenerative models, including inhibition of GSK-3β, pathogenic protein formation, and GSK-3β and Aβ pathogenic effects. These drugs have also improved mitochondrial energy production and energy preservation after injury and protected against mitochondrial depolarization, transition pore development, and cytochrome c release. Additionally, these agents have reduced free radical production and lipid peroxidation, particularly free radicals induced by Aβ, and inhibited apoptosis, especially that induced by glutamate, Aβ, MPP +, and the proteasome inhibitor MG132 (relevant to Huntington’s disease, amyotrophic lateral sclerosis, Alzheimer’s disease, Parkinson’s disease, and other neurodegenerative diseases). Finally, SNRI properties of ziprasidone further suggest the capacity to reduce GSK-3β, pathogenic protein concentrations, nitric oxide synthesis, other free radicals, mitochondrial depolarization, apoptosis, microglial activation, and neuroinflammation while upregulating BDNF and GDNF.
In considering the putative neuroprotective effects of ziprasidone, we will sequentially consider the neuroprotective effects associated with D2 antagonists, drugs with 5HT2 antagonist properties, and 5HT1a agonists (SNRI properties are described in the duloxetine section).
D2 antagonists inhibit Aβ formation,
272 Aβ-induced calcium imbalances,
273 and apoptosis induced by glutamate in Huntington’s disease transgenic mice,
227 MPP
+ in a Parkinson’s disease model,
274,
275 and MG132 proteasome inhibition in PC12 cells.
276 These actions are mediated through antipsychotic effects on D2 receptors,
273 sigma receptors,
272,
273 respiratory chain complex I and II inhibition,
276 and mitochondrial transition pore development.
274,
275Ziprasidone and other D2-5HT2 antagonist antipsychotics have inhibited GSK-3,
212 leading to reductions in Aβ, tau, αSyn, and mitochondrial dysfunction (see introduction). 5HT2 receptor stimulation regulates GSK-3β activity by reducing serine 9 phosphorylation,
214 indicating that 5HT2 antagonism can promote GSK-3β inhibition. D2-5HT2 antagonists have increased energy charge and redox reaction velocity,
277 increased ATP production,
278 and prevented ATP reductions after injury.
279,
280 They also have reduced superoxide radicals,
281 protected against Aβ-induced reactive oxygen species,
282 and inhibited mitochondrial depolarization,
282 mitochondrial permeability transition pore development,
281 and cytochrome c release.
278,
281 Ziprasidone has been shown to reduce lipid peroxidation.
283 Finally, D2-5HT2 antagonists exhibit antiapoptotic properties including calmodulin antagonism
284 and endonuclease inhibition
284 and have inhibited apoptotic events in a variety of models,
284 –
289 including Aβ-induced
282,
290 and MPP
+ -induced
291 apoptosis in neuronal PC12 cells.
In light of its SNRI properties, ziprasidone may have the additional antiapoptotic property of monoamine oxidase B inhibition.
292 5HT1a agonists have also demonstrated antiapoptotic effects.
293 –
296 Moreover, 5HT1a receptor stimulation also inhibits GSK-3β through serine 9 phosphorylation.
214The combined D2 and 5HT2 antagonist, 5HT1a agonist, and SNRI properties of ziprasidone suggest a wealth of potential neuroprotective actions for this agent. The potential neuroprotective actions of ziprasidone are summarized in
Table 2 .
S-Zopiclone
S-zopiclone is a short half-life nonbenzodiazepine cyclopyrrolone GABA-A receptor subunit agonist with preference for omega-1 receptors.
297 Short half-life benzodiazepine receptor site-specific agonists such as s-zopiclone are considered first-line treatments for sleep disturbances in Alzheimer’s disease
298,
299 and Parkinson’s disease.
16 Short half-life hypnotics have been found to be safe and effective without notable side effects in the elderly,
300 and zopiclone is particularly well tolerated
301 and effective.
302S-zopiclone and zopiclone have each been demonstrated to improve anxiety, depression, and cognitive impairment in patients with insomnia.
303 S-zopiclone REM suppression
304 may also improve REM behavior disorder in synucleinopathic neurodegenerative diseases. Moreover, “sleep benefit” has been observed for Parkinson’s disease motor symptoms.
305 –
308 Similarly, sleep appears to replenish neurotransmitter availability in dementia, especially acetylcholine,
309 the depletion of which has been linked to behavioral
310,
311 and cognitive
312 disturbances. Consistent with this concept, sleep disturbances are correlated with reduced function in Alzheimer’s disease and in nondemented elderly.
313 A number of neuropsychiatric features are sensitive to sleep, including cognitive and noncognitive conditions. Noncognitive neuropsychiatric conditions that are sensitive to sleep include depression,
305 anxiety,
314 perceptual distortions and hallucinations,
315,
316 aggressive behavior,
317 and apathy.
318 Sleep-sensitive cognitive conditions include delirium,
319 cognitive impairment,
318 psychomotor impairment,
320 attention and vigilance,
321 –
325 working memory,
326 –
331 learning,
332,
333 short-term recognition memory encoding,
334 semantic memory retrieval,
326 hippocampus-dependent declarative memory,
335,
336 visual memory,
325,
337 visual processing,
325 long-term memory,
338 frontal executive performance,
309,
326,
339 –
342 response inhibition,
343 reasoning,
344 and other higher cognitive functions.
345Zopiclone and similar agents have improved insomnia and nocturnal wandering,
346 anxiety,
347,
348 and agitation
298,
349 –
353 arising within the context of dementia. These drugs have also been reported to improve aggression in brain disease,
354,
355 psychosis,
356 –
358 depression,
359,
360 and apathy in schizophrenia.
361,
362 Thus, s-zopiclone has the potential to improve a wide range of neuropsychiatric, cognitive, and motor disturbances in patients with neurodegenerative diseases.
Although the effects of s-zopiclone on neurodegenerative disease pathophysiology are in the early stages of investigation, s-zopiclone and other drugs sharing properties of s-zopiclone seem to exhibit neuroprotective properties that include protection against pathogenic proteins, free radical-induced lipid peroxidation, mitochondrial transition pore development, cytochrome c release, and apoptosis. There is also evidence of neurorestorative effects on GSK-3β over-activity, proteasomal processing, hippocampal neuronal ATP production, neuroinflammation, and BDNF expression.
GABA-A agonists have been shown to protect against Aβ-induced neurotoxicity,
363 glutamate-induced free radical formation,
364 free radical-induced lipid peroxidation,
365 calcium-induced mitochondrial permeability transition pore development,
366 and ischemia-induced mitochondrial cytochrome c release. They also promote ATP recovery in ischemic hippocampal slices
367 and may protect against apoptosis induced by Aβ
363 and by glutamate in hippocampal neurons.
364 These antiapoptotic properties are in contrast to GABA-A agonists that bind to peripheral benzodiazepine (TSPO) receptors (e.g., diazepam) and promote apoptosis. Finally, sleep deprivation, often present in neurodegenerative diseases, has been associated with GSK-3β activation,
368 altered proteasomal processing,
369 oxidative damage,
370 impaired mitochondrial integrity and function,
371 neurodegenerative inflammation,
102 and decreased BDNF expression.
372 (While sleep deprivation increases BDNF in developing rats,
373 developing tissue and organisms are different from mature tissues and organisms.) Correction of the insomnia with treatment can potentially reverse these impairments. S-zopiclone and related drugs may therefore provide polymodal neuroprotection in these diseases. The potential neuroprotective actions of s-zopiclone are summarized in
Table 2 .
Ramelteon
Ramelteon is a melatonin (MT) MT1 and MT2 receptor agonist
374 with demonstrated efficacy in elderly subjects with insomnia.
375,
376 Ramelteon is more potent and has greater affinity at melatonin receptors than does melatonin itself.
374 As mentioned above (see s-zopiclone section, third paragraph), a large number of neuropsychiatric features are sensitive to sleep. Additionally, reduced melatonin excretion has been observed in beta-blocker hallucinosis,
377 a condition that resembles the hallucinations
378 and delirial features
379 of Parkinson’s disease and dementia with Lewy bodies. Consistent with these symptoms induced by beta-blockers, noradrenergic locus coeruleus degeneration is observed in Parkinson’s disease
380 and dementia with Lewy bodies,
381 suggesting a potential link between these symptoms and melatonin receptor activity.
Melatonin administration itself has improved sleep
382,
383 and has demonstrated efficacy in neuropsychiatric disturbances in general,
383 including anxiety,
384 delirium,
385,
386 disturbances of attention,
387,
388 some aspects of memory,
384,
389 daytime sleepiness and “sundowning behavior” in dementia,
386 and REM behavior disorder
390 that is commonly seen in dementia with Lewy bodies and Parkinson’s disease dementia.
15,
16 Melatonin has also been suggested as a treatment for fluctuations in cognition and alertness in Parkinson’s disease.
391 The greater potency and affinity of ramelteon at melatonin receptors may lead to outcomes superior to those obtained with melatonin.
Melatonin has many neuroprotective actions in preclinical models, as exemplified in Parkinson’s disease and Alzheimer’s disease. As mentioned in the s-Zopiclone section, sleep deprivation promotes neurodegenerative disease through effects on GSK-3β, the proteasome, oxidative damage, mitochondrial impairment, and neuroinflammation. Besides improvement in these functions ascribable to alleviating insomnia in neurodegenerative disease, results of melatonin treatment studies suggest ramelteon may exert neuroprotective effects by preserving dopaminergic,
392 –
402 striatal,
396,
399,
403 –
406 and hippocampal
403 neurons. Other potential neuroprotective actions include inhibiting αSyn aggregation,
407,
408 pathogenic tau formation and tau-induced pathogenesis,
282,
409 –
419 and Aβ fiber formation, aggregation, and deposition
411,
412,
420 –
422 and preventing tau- and Aβ-induced αSyn oligomerization.
112 –
114,
116 Moreover, ramelteon has the potential to inhibit nitric oxide synthase,
423 scavenge free radicals,
396,
397,
403,
404,
423 –
429 induce antioxidant enzymes,
423,
430 prevent dopamine auto-oxidation,
431 –
434 maintain mitochondrial integrity,
425,
435 –
438 and protect against losses of complex I
439 –
446 in Parkinson’s disease and complex IV.
440,
442 –
447 Furthermore, ramelteon may be able to prevent apoptotic cascades,
395,
397,
399 –
402,
404 –
407,
424,
428,
430,
448 –
456 particularly those induced by glutamate,
457 –
459 Aβ,
460 and MPP
+ 406,
454,
456,
461 apoptosis. It is particularly interesting to note that the protection afforded against glutamate-induced apoptosis appears to be mediated by activation of the MT1 receptor.
459 Finally, ramelteon may stimulate BDNF,
462 –
464 GDNF,
108,
110,
462 –
465 and neuritogenesis.
103 Each of these effects is particularly relevant to neurodegenerative disease pathogenesis (see introduction), and ramelteon may prove to clinically protect against neurodegeneration.
The evidence is far too extensive to review here (see Part I of this report, and Table 3 of the online data supplement to Part I of this report), but by way of examples in Parkinson’s disease, melatonin prevented αSyn aggregation after maneb
407 and rotenone exposure
408 in Parkinson’s disease models and has preserved dopaminergic tyrosine hydroxylase-positive nigral neurons bearing MT1 and MT2 receptors after 6-hydroxy-dopamine lesions in rats.
395 Melatonin has prevented nigrostriatal free radical damage in mice
403 and rats
396 treated with MPTP and has led to essentially full nigrostriatal recovery in rats treated with MPTP
397 and those treated with 6-hydroxydopamine.
404 Melatonin reduced auto-oxidative semiquinone formation and increased
l -dopa bioavailability in rats treated with intrastriatal
l -dopa.
433 A rat striatal 6-hydroxydopamine study indicated neuroprotection by melatonin manifest in reduced nigral neurodegeneration as evidenced by apomorphine-induced turning behavior.
405 Melatonin reduced nigrostriatal neuronal apoptosis in an MPTP mouse model.
406 BDNF is colocalized with dopamine neurons in the substantia nigra, where it may exert neuromodulatory and neuroprotective effects.
244 BDNF and GDNF correlate with MT1 receptor activity,
463 and melatonin increases both GDNF and BDNF and promotes the viability of MT1 and MT2 receptor-bearing neural stem cells derived from rat ventral midbrain. It also significantly increases tyrosine hydroxylase and its mRNA,
464 indicative of enhanced nigral dopaminergic neurotrophism that is highly relevant to Parkinson’s disease. Melatonin and dopamine exhibit similar gene expression and protein profiles.
452,
453 Recently, ramelteon has been demonstrated to increase neuronal BDNF concentrations in cultured mouse cerebellar granule cells bearing MT1 and others bearing MT2 receptors.
466Neither melatonin nor ramelteon has undergone clinical trials for its clinical and disease-modifying neuroprotective properties in neurodegenerative disease, and melatonin preclinical and ramelteon clinical findings remain to be confirmed in neurodegenerative disease patients. An important caveat regards the need to determine which of the above neuroprotective properties of melatonin are mediated through melatonin receptors and other mechanisms common to both melatonin and ramelteon. The potential neuroprotective actions of ramelteon are summarized in
Table 2 .
Directions for Future Research
The potential neuroprotective actions for each agent have been specified above. In
Table 1 (also discussed in Part I of this report), we have indicated candidate criteria for potential prediction of translational neuroprotection in clinical patients.
1.
We therefore suggest that each of these promising agents be evaluated in terms of the specific enumerated neuroprotective actions in accord with the criteria of
Table 1 . Such inquiries can clarify the mechanisms of these drugs and may determine the likelihood of positive outcomes in randomized, double-blind, placebo-controlled delayed-start or randomized-withdrawal neuroprotective clinical trials.
125 Although ethical issues regarding withholding treatment have been raised in such trials, it cannot be known whether a given drug
has neuroprotective efficacy
until such trials are conducted. In evaluating antidepressants in patients with depression, these procedures might be modified by using an antidepressant with less robust neuroprotective properties to supplant the placebo, avoiding an ethical dilemma by providing equally efficacious treatment for the depression.
2.
Furthermore, given that these agents are widely used to treat clinical neuropsychiatric conditions that arise in the context of neurodegenerative disease, and given that their safety and tolerability are known, neuroprotective clinical trials need not necessarily await the fulfillment of the
Table 1 criteria. Nevertheless, caution is advised in light of adverse events recently identified for psychotropic drugs (e.g., increased death and stroke risks with antipsychotics) in elderly populations, the population most at risk for the most common neurodegenerative diseases.
3.
Research should be continued into the correlation of psychotropic pharmacodynamic mechanisms with neuroprotective actions.
4.
Finally, research to determine the clinical neuroprotective predictive utility of each of the
Table 1 criteria can greatly assist the efforts of propelling agents of interest from preclinical studies to the clinical trial stage.