Alzheimer's disease is the most common cause of dementia in the elderly, affecting about 10% of people older than 65 years (
1). Years before diagnosis, patients suffer from progressive cognitive impairment, suggesting the existence of pathological processes in early preclinical stages of the disease. Therefore, it is essential to identify risk factors that contribute to structural and functional brain changes associated with or preceding memory loss. The e4 allele of the apolipoprotein E gene (APOE-4), the only well-established genetic risk factor for late-onset sporadic Alzheimer's disease (
2), is associated with structural (
3) and functional (
4) brain changes early in life. The risk of developing Alzheimer's disease also increases with a positive first-degree family history of the disease (
5,
6). There has been controversy surrounding whether this effect is independent from APOE-4 risk (
7), which is itself associated with familial aggregation of Alzheimer's disease (
8). It is also unknown which genetic or nongenetic mechanisms determine a family history. APOE-4 and family history risks highly co-occur (
9), and they possibly interact (
10). The preferred clinical strategy is to investigate multiple risk factors and to establish diagnostic or therapeutic decisions based on risk factor patterns. Therefore, it is essential to understand how and how specific these risk factors contribute to Alzheimer's disease-related pathology.
Whereas APOE-4-associated differences in cognitive abilities or in brain structure and function are increasingly studied, little is known about independent effects of a family history of Alzheimer's disease on cognitive functioning, and neuroimaging data are rare. La Rue et al. (
11) found an exaggerated recency effect in word list learning among cognitively normal middle-aged subjects with a family history of Alzheimer's disease. This serial position pattern, which can be found in Alzheimer's disease patients, suggested greater reliance on immediate memory in subjects with the family history risk factor relative to subjects without this risk factor. The effect was independent of the participants' APOE genotype.
Subjects with maternal family history of Alzheimer's disease show reduced glucose metabolism with positron emission tomography in brain areas susceptible to the disease pathology, when compared with subjects with paternal or no family history (
12). Additional longitudinal investigation revealed that maternal family history of Alzheimer's disease was also associated with more rapid metabolic decline (
13). Using functional magnetic resonance imaging (fMRI) during an encoding task, Johnson et al. (
14) and Trivedi et al. (
15) found reduced medial temporal lobe activity among APOE-4 carriers with a family history of Alzheimer's disease. Bookheimer et al. (
16) showed greater hippocampal activations in APOE-4 carriers relative to noncarriers during memory tasks. Johnson et al. (
14) suggested that modeling family history of Alzheimer's disease may help to explain discrepancies among fMRI studies investigating subjects at APOE-4 genetic risk.
Among siblings discordant for Alzheimer's disease, the heritability for cerebral or medial temporal lobe atrophy and white matter lesions is high and cannot be explained by APOE status alone (
17). Structural magnetic resonance imaging (MRI) measures can reveal anatomical traits that could be associated with cognitive impairment or may help to predict future conversion to Alzheimer's disease. Decreased hippocampal volume is the most robust MRI marker for imminent conversion to Alzheimer's disease in subjects with mild cognitive impairment (
18). The earliest neuropathological features of Alzheimer's disease arise from the medial temporal lobe, particularly the entorhinal region (
19). Therefore, in the analysis of cognitively healthy subjects at APOE-4 genetic risk, MRI investigations have been focused mainly on the hippocampus and entorhinal cortex. Studies investigating the association between the APOE-4 genotype and hippocampal volume have produced mixed results (
20–22). We previously reported cortical thickness differences among medial temporal lobe subregions associated with APOE genotype in cognitively normal elderly subjects. Further, cortical thickness measures of medial temporal lobe subregions were more sensitive than hippocampal volumetry (
23).
It is unknown how APOE-4 risk and family history risk individually contribute to structural brain differences in healthy individuals, whether the effects are overlapping, additive, or interacting. Investigating brain structure in cognitively healthy people at risk rather than patients diagnosed with Alzheimer's disease makes it possible to identify the contribution of individual risk factors that might be obscured by extensive brain atrophy once the disease begins. Because of the expected subtlety of differences among cognitively normal subjects, we performed cortical unfolding using high-resolution MRI (
24,
25), separately measuring cortical thickness among subregions of the medial temporal lobe. We hypothesized that a family history of Alzheimer's disease would be associated with a thinner cortex in regions known to be affected early in the disease, specifically the entorhinal cortex and hippo-campal subfields, after accounting for APOE genotype. Further, we hypothesized that the effects of APOE-4 and family history risk would be additive in predicting hippo-campal atrophy.
Method
Subjects
Fifty-one subjects (mean age: 62.3 years [SD=10.7]; range=38-86 years; 26 subjects with and 25 without a family history of Alzheimer's disease) underwent APOE genotyping (
26), MRI scanning, and neuropsychological assessments from 2004 to 2009. Participants were selected from a pool of 230 subjects recruited through advertisements. We excluded subjects with a history of psychiatric or neurological disorder or a major systemic disease affecting brain function. We also excluded individuals who were not cognitively normal, according to National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer's Disease and Related Disorders Association criteria (
27); scored <27 points on the Mini-Mental State Examination (
28); or scored >0 points on the Clinical Dementia Rating Scale (
29). Cognitive status was further assessed with additional standardized tests (
Table 1) according to our standard battery (
30), which includes measures for memory (Wechsler Memory Scale, logical memory/ delayed recall and verbal paired associations II; Buschke-Fuld Selective Reminding Test, consistent long-term retrieval; Rey-Osterrieth Complex Figure, delayed recall) and other major cognitive domains, such as language, attention/executive functioning, and visuospatial skills (Boston Naming Test; FAS Test; Wechsler Adult Intelligence Scale, digit span; Stroop Interference; Wisconsin Card Sorting Test; Trailmaking Test, Part B; Rey-Osterrieth Complex Figure, copy). Four subjects were excluded because they did not give consent to APOE genotyping, and we excluded one subject with an APOE 2/4 genotype.
Investigators performing scanning and cortical unfolding procedures were unaware of the participants' APOE and family history status. Family history of Alzheimer's disease was considered positive if at least one first-degree relative had been diagnosed with the disease using standard criteria (
27). In one case, diagnosis was confirmed by autopsy. All participants with a family history had parental family history only. After complete description of the study to the subjects, written informed consent was obtained in accordance with the UCLA Human Subjects Protection Committee procedures.
Procedures
MRI scanning was performed on a Siemens Allegra 3-Tesla whole brain MRI scanner (Siemens Medical Solutions, Inc., Malvern, Pa). T1-weighted magnetization-prepared rapid acquisition gradient-echo scans were acquired (repetition time: 2,300 msec; echo time: 2.93 msec; slice thickness: 1 mm; 160 slices; in-plane voxel size: 1.3×1.3 mm; field of view: 256 mm) for volumetric measurements, and high-resolution oblique coronal T2-weighted fast-spin echo sequences were acquired for structural segmentation and unfolding procedures (repetition time: 5,200 msec; echo time: 105 msec; slice thickness: 3 mm; spacing: 0 mm; 19 slices; in-plane voxel size: 0.39×0.39 mm; field of view: 200 mm).
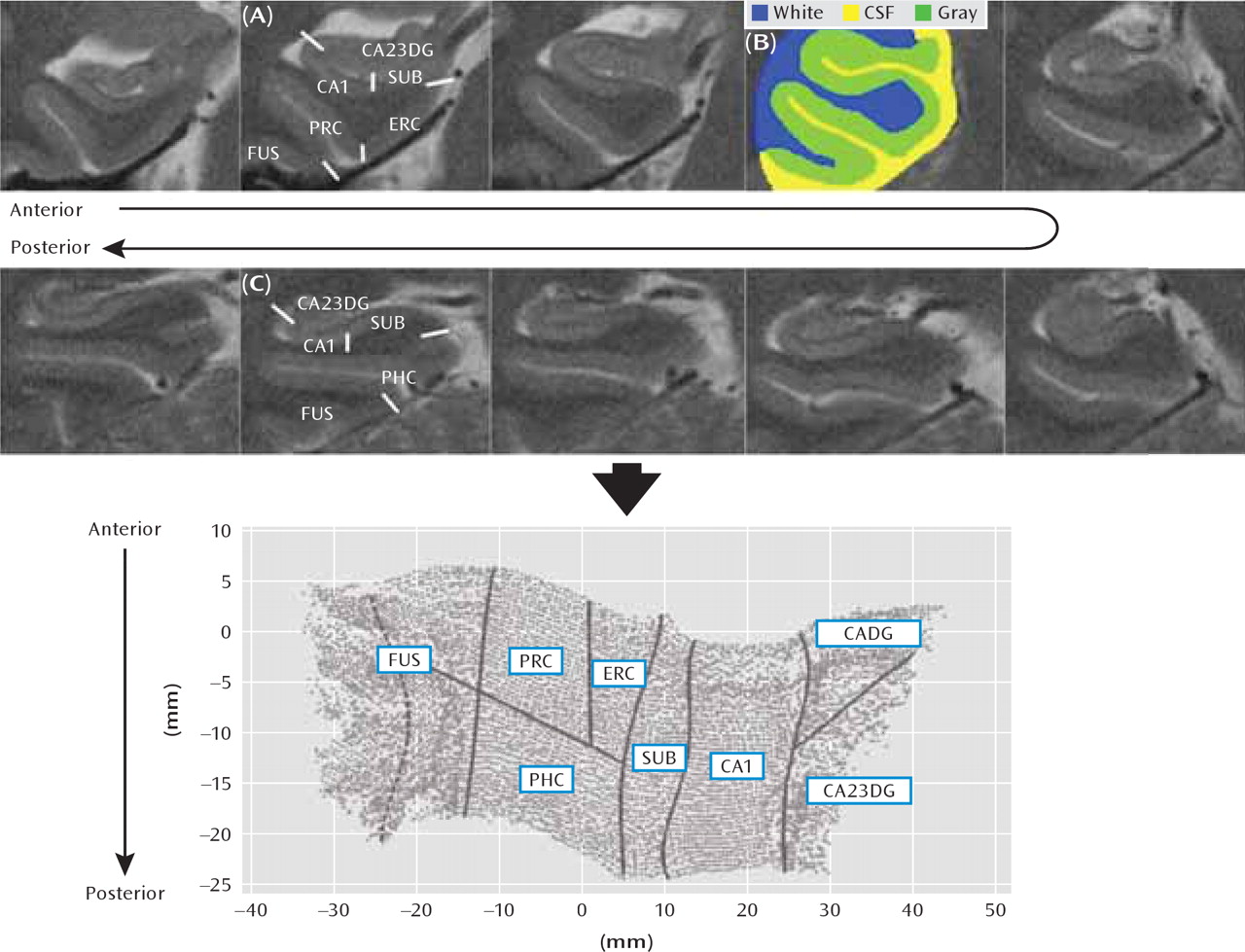
Cortical unfolding enhances the visibility of the convoluted medial temporal lobe cortex by flattening the entire gray matter volume into two-dimensional space (
Figure 1). This procedure is detailed elsewhere (
24,
25). Briefly, gray matter is specified by manually defining white matter and CSF on coronal fast-spin echo MRI data. To improve segmentation, the original images are interpolated by a factor of seven. Then, up to 18 connected layers of gray matter are grown out using a region-expansion algorithm to cover all pixels defined as gray matter, resulting in a gray-matter strip containing cornu ammonis fields 1–3, the dentate gyrus, the subiculum, entorhinal, perirhinal, and parahippocampal cortices, and the fusiform gyrus. Boundaries between subregions were delineated on the original in-plane MRI images, based on histological and MRI atlases, and then mathematically projected to their flat map space coordinates. Accuracy of the unfolding procedure, resulting in topographically correct flat maps, has been shown extensively (
23–25). Cortical thickness was calculated in three-dimensional space in all subregions. For each gray matter voxel, the distance to the closest nongray matter voxel was computed. In two-dimensional space, for each voxel, the maximum distance value of the corresponding three-dimensional voxels across all layers was taken and multiplied by two. The mean thickness in each subregion was calculated by averaging the thickness of all two-dimensional voxels within each region of interest. Global hippocampal thickness was computed by averaging across all subregions.
Comparison of brain volumes between subjects was performed using Functional MRI of the Brain Software Library (
http://www.fmrib.ox.ac.uk/fsl/), with T1-weighted magnetization-prepared rapid acquisition gradient-echo scans. Medial temporal lobe volumes were obtained by averaging the volume of each unfolded region's voxels in three-dimensional space (
23). We measured brain volumes from the T1-weighted scans rather than using high in-plane resolution T2-weighted data, since these images are easily affected by subtle differences in slice orientation and field of view. Both whole brain volume-corrected and raw cortical thickness data were analyzed. Volume correction was performed by multiplying each subject's cortical thickness values with an individual correction factor, derived from the average volume for all subjects divided by the individual subjects' volume. We then conducted the statistical analyses as described below. No difference occurred in the pattern of significant results due to brain volume correction. Thus, we always refer to raw data in our analyses.
Statistical Analyses
To determine whether APOE-4 genotype and family history of Alzheimer's disease are associated with a thinner cortex, we first estimated regression models, using global (averaged across subregions) hippocampal thickness as the dependent variable and genotype, family history, and age as predictors. Interactions between these predictors were also examined. We then calculated the percent of variance in cortical thickness explained by each predictor.
In order to ascertain regional differences, we estimated mixed general linear models, with genotype and family history as between-group factors and age as a covariate. Because we were investigating multiple subregions, we used subregions as a within-group factor and conducted post hoc univariate tests only after significance was established by the multivariate F tests in order to determine the subregions that contributed to the significant findings. Thus, this method served as a safeguard against spurious findings. Additional analyses of variance were performed for medial temporal lobe and brain volume comparisons as well as analyses of neuropsychological data. Statistical analyses used a significance level of p<0.05.
Results
There were no significant differences in neuropsycho-logical test scores between subjects with APOE and family history risk factors (
Table 1). Subjects did not significantly differ in educational status, mean age, and age distribution. No difference in medial temporal lobe or overall brain volume could be detected between the groups. Additionally, there was no correlation between neuropsychological test performance and hippocampal thickness.
The regression model for global hippocampal cortical thickness revealed significant contributions for family history of Alzheimer's disease (F=11.04, df=1, 47, p=0.002) and genotype (F=7.72, df=1, 47, p=0.01) but not for age. None of the interaction terms was significant. Age, while not significant, explained approximately 5.1% of the variance, genotype approximately 13.7%, and family history approximately 17.8% if investigating the factors separately. Given both age and genotype, family history explained another 15.9% of the variance. Given both age and family history, genotype explained another 11.2% of the variance.
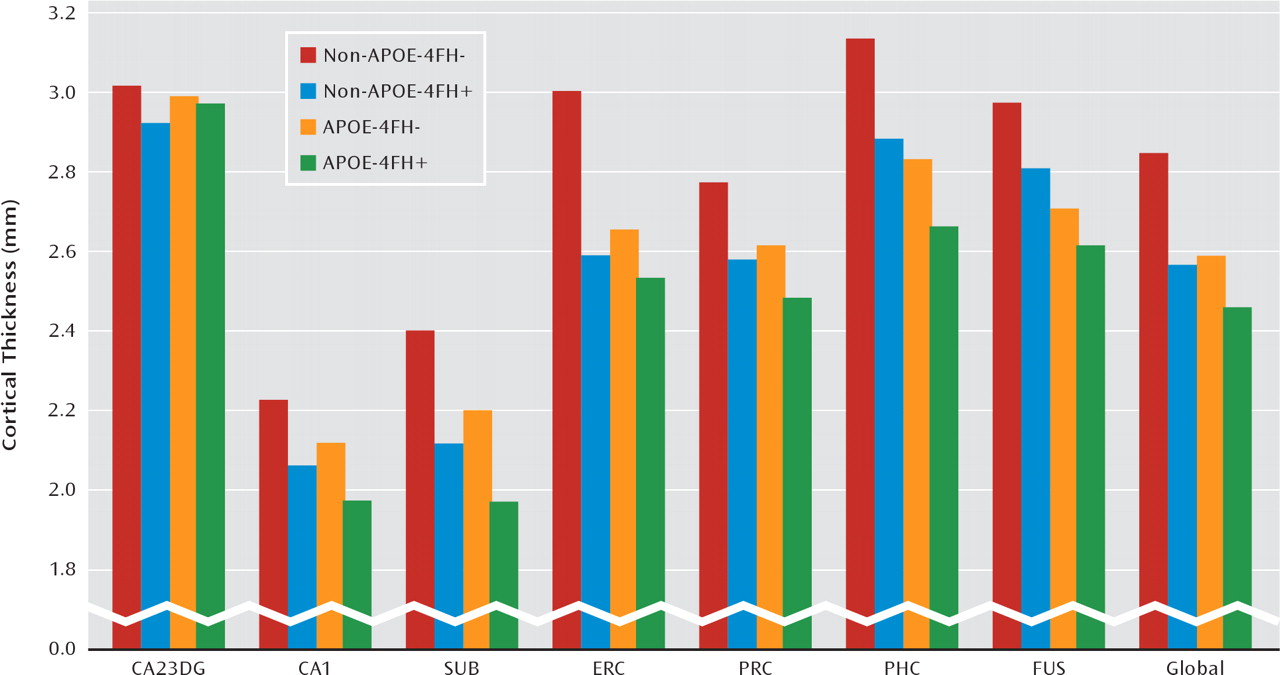
The results of cortical thickness analyses are shown in
Figure 2. The mixed general linear models yielded significant main effects for family history (F=8.74, df=1, 47, p=0.005) and genotype (F=5.38, df=1, 47, p=0.025) as well as subregion-by-APOE interaction (F=2.62, df=6, 44, p=0.029) and subregion-by-family history interaction (F=2.37, df=6, 44, p=0.046). The main effect for age did not reach significance. Other possible interactions were not significant. Subjects with a family history of Alzheimer's disease when compared with subjects without this risk factor showed a significant thinner cortex in cornu ammo-nis field 1 (p=0.001), the subiculum (p=0.001), the entorhinal cortex (p=0.003), and the parahippocampal cortex (p=0.023). APOE-4 allele carriers compared with noncarriers showed a significant thinner cortex in the subiculum (p=0.033), the entorhinal cortex (p=0.03), the parahippocampal cortex (p=0.005), and the fusiform gyrus (p=0.001).
Across the medial temporal lobe as a whole, cortical thickness was reduced approximately 7% for subjects who had either an APOE-4 risk or a family history risk but was reduced 13% for those with both risk factors relative to subjects with neither risk factor. This approximately additive pattern could not be found in the entorhinal cortex, where having two risk factors produced about the same effect as having either an APOE-4 risk or a family history risk (
Figure 2).
Discussion
This study demonstrates that among cognitively normal subjects, a family history of Alzheimer's disease and the APOE-4 genotype contribute independently and additively to cortical thinning in the hippocampal region. An additive effect of family history and APOE genotype could not be found in the entorhinal cortex, where either risk factor produced decreased cortical thickness, but a second risk factor did not alter the degree of atrophy. Family history accounted for a greater percent of the unique variance in global hippocampal thickness as explained by the APOE genotype and was associated with a thinner cortex in the hippocampal cornu ammonis field 1, subiculum, entorhinal cortex, and parahippocampal cortex. In line with our previous results (
23), APOE-4 carriers, when compared with noncarriers, showed a thinner subiculum and entorhinal cortex, and we additionally detected a thinner parahippocampal cortex and fusiform gyrus.
Despite the fact that family history of Alzheimer's disease and APOE genotype contributed independently to the variance in cortical thickness, they affected the same cortical regions, which are those reported to show the earliest signs of the disease-related pathology (
19). For clinicians, it is important to notice that the family history risk factor contributes substantially to structural brain differences within the medial temporal lobe among healthy relatives of Alzheimer's disease patients, highlighting the significance of the diagnosis for these relatives. For researchers, it could be critically important to control for family history effects in studies investigating other Alzheimer's disease risk factors (
14).
It has been hypothesized that there are yet unknown susceptibility genes for late-onset Alzheimer's disease of perhaps equal or larger effect size relative to that for APOE-4 (
31). We can assume that these unknown genetic factors would be represented in family history risk. The co-occurrence and overlap of both risks could mask APOE-4-related contributions that could or could not differ from family history effects. Carrying the APOE-4 allele increases the risk of developing Alzheimer's disease and affects the onset, but perhaps not the progress, of the disease (
32,
33) and remains an important factor throughout the lifespan (
34). Family history also increases the risk of developing the disease (
5), but it is not known how the risk factor influences disease progression or if the effects vary with an individual's age. Studies investigating these family history effects did not account for APOE status (
35,
36). It is further controversial whether both risk factors interact. van Duijn et al. (10 and the authors' response to 37) found that family history was associated with increased risk in heterozygous but not homozygous APOE-4 carriers. Others emphasize additive effects of APOE-4 and family history risks on the prediction of the disease (
37). Although we found APOE-related effects in the subiculum and entorhinal cortex, possibly reflecting these regions' susceptibility to early Alzheimer's disease-related pathology (
19), family history was associated with a thinner cortex also in the adjacent cornu ammonis field 1. The patterns of cortical thickness among medial temporal lobe subregions associated with family history risk or APOE-4 risk could reflect at least partially different mechanisms contributing to cortical atrophy. It has been shown that family history of Alzheimer's disease is associated with both whole brain and medial temporal lobe cortical atrophy as well as white matter lesions (
17).
Differential effects of age, APOE-4 carrier status, and Alzheimer's disease on hippocampal subfields have been reported previously. Mueller and Weiner (
22) found reduced volume in hippocampal cornu ammonis fields 1 and 3 and the dentate gyrus associated with older age, whereas APOE-4 carrier status was associated with reduced cornu ammo-nis field 3 and dentate gyrus volume only. Dickerson et al. (
38) revealed moderately reduced volume in the entorhinal and posterior parahippocampal cortex among healthy elderly individuals relative to younger individuals. Differences between these findings and our study may reflect that most studies examine volume measures. Volume reduction may occur because of reduced cortical surface area rather than cortical thinning among healthy elderly subjects (
38).
There are several limitations to this study. Our study was not primarily aimed at investigating age effects on hippo-campal cortical thickness. Therefore, this could be studied more directly with a larger number of participants, balanced across different age ranges. Although based on standard clinical criteria, it is possible that relatives with dementia other than Alzheimer's disease were included. Furthermore, healthy relatives could develop Alzheimer's disease in the future, and the presumably heterogeneous pattern of factors contributing to family history could differ among subjects. Finally, in APOE-4 subjects with a family history, it is not possible to determine clinically whether two different risk factors are present, since these participants could have a family history because of the APOE-4 allele.
The detection of structural brain changes in preclinical stages of Alzheimer's disease and in subjects at risk for the disease using MRI is becoming increasingly important and focuses on brain areas that are affected earliest in the disease (
18,
39). The possibility to identify macroscopic brain structure differences associated with Alzheimer's disease risk factors in healthy individuals could point to the future use of MRI in aging. This could range from diagnostically monitoring the dynamics of these changes to measuring effects of strategies aimed to delay or prevent cognitive decline (
39).
The risk for developing Alzheimer's disease can be modulated by various and rarely studied factors, ranging from yet unidentified genes and gene-gene interactions to environmental/lifestyle contributions (
40). Neither the APOE-4 genotype nor a family history of Alzheimer's disease are sufficient to cause the disease. Therefore, the pattern of risk factors and their relative predictive value have strong implications for a physician's decision on how to educate, monitor, and treat an individual. We show that family history modulates brain structure independently and additively to APOE-4-associated effects in brain regions most susceptible to the disease pathology and accounts for a greater percent of the variance in global hippocampal thickness than APOE-4 risk. Follow-up investigations will be necessary to determine which individuals will ultimately develop Alzheimer's disease. Our findings underline the importance of a family history risk factor in clinical evaluations as well as imaging genetics in people at risk for Alzheimer's disease.
Acknowledgments
The authors thank Ms. Andrea Kaplan and Ms. Debbie Dorsey for assistance in subject recruitment, data management, and study coordination.