Brain-derived neurotrophic factor (BDNF) is widely distributed throughout the adult brain. BDNF binds to the nonselective p75 receptor, common to a variety of neurotrophins, as well as to its specific high-affinity TrkB receptor. There is evidence that BDNF plays a critical role in long-term potentiation (
1) and is a molecular mediator of learning and memory. Intrahippocampal infusion of BDNF in vivo and BDNF infusion in hippocampal slices induce long-lasting enhancement of synaptic plasticity (
2–4). In addition, BDNF has been shown to be involved in declarative and spatial memory processes (
5,
6). For these reasons, BDNF is an attractive candidate for modulating learning and memory processes.
Deprivation of endogenous BDNF causes impairment of spatial learning and memory in rats (
7), and forebrain-restricted deletion of BDNF severely impairs spatial learning (
8). Similarly, BDNF and TrkB are both known to play an important role in fear memory acquisition and extinction (
9,
10). We previously demonstrated that disruption of TrkB activation using lentiviral expression of a dominant-negative form of TrkB into the basolateral amygdala blocked the acquisition of fear (
11) and the consolidation of extinction (
12), which suggests that BDNF-dependent activation of TrkB in the amygdala regulates the learning of fear and extinction memories.
Recent studies indicate that BDNF polymorphisms may be implicated in emotionality and anxiety disorders (
13,
14). In addition to the role of the BDNF/TrkB system in learning, memory, and synaptic plasticity, alterations within the system have been implicated in affective disorders (
15). Despite this increasing awareness of the many important roles played by BDNF activation of TrkB, a fuller understanding of this system and the use of potential TrkB-acting therapeutic agents has been limited by the lack of any identified small-molecule TrkB agonists that fully mimic the actions of BDNF at brain TrkB receptors in vivo.
Jang et al. (
16) recently screened a chemical library for compounds that activate TrkB in vitro and discovered that a series of flavone derivatives, most potently 7,8-dihydroxyflavone (7,8-DHF), significantly activated TrkB. They found that 7,8-DHF binds in vitro with high affinity to the TrkB receptor and provokes its dimerization and autophosphorylation, leading to downstream signaling cascade activation. Systemic administration of 7,8-DHF protects neurons in wild-type but not TrkB-deficient or mutated (F616A) mice from apoptosis, substantially activates TrkB in the brain 2 hours after injection, inhibits kainic acid-induced neuronal cell death, decreases infarct volumes in stroke in a TrkB-dependent manner, is neuroprotective in an animal model of Parkinson's disease (
16), and rescues a BDNF deficit in a BDNF cortex-specific knockout murine model (
17). To more fully examine the effects of this newly identified TrkB agonist, we examined the effects of 7,8-DHF on the acquisition and extinction of cue-dependent conditioned fear among the most simple yet robust rodent models of emotional learning and memory.
Method
Animals
All experiments were performed on adult (2-4 months old) wild-type strain C57BL/6J male mice from Jackson Laboratory (Bar Harbor, Me.). All procedures were approved by the Institutional Animal Care and Use Committee of Emory University and were in compliance with National Institutes of Health guidelines. Separate cohorts of animals were used in each experiment.
Drugs
We administered 7,8-DHF (obtained both from coauthor K.Y. and from Tokyo Chemical Industry, catalog no. D1916) intraperitoneally at a dose of 5 mg/kg in a vehicle of 17% dimethylsulfoxide in phosphate-buffered saline; the same vehicle was also used in control groups. In those experiments in which 7,8-DHF was given, mice received a single dose 1 hour before the appropriate behavioral procedure.
Immunoblotting, Immunohistochemistry, and Autoradiography
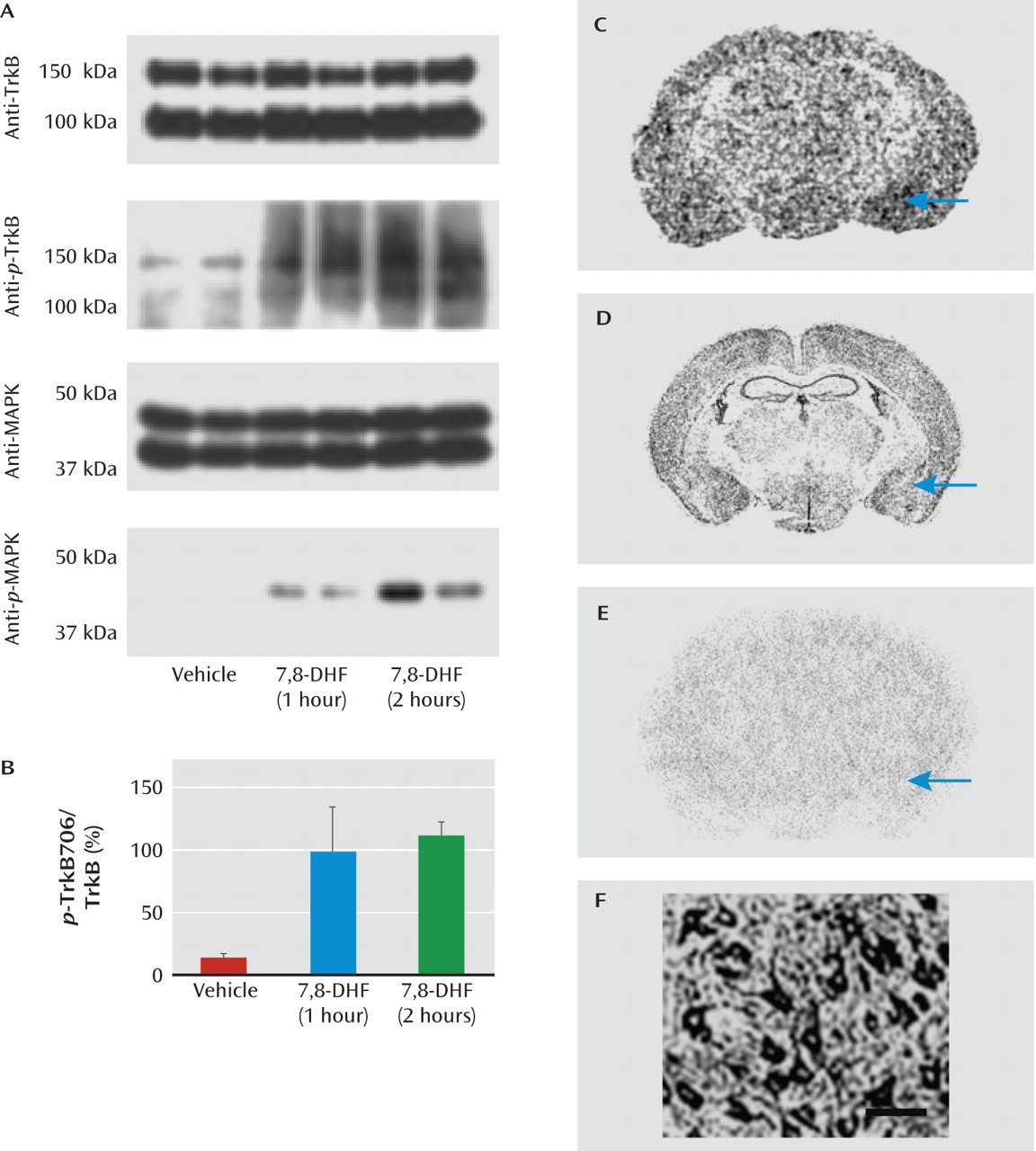
For Western blots, mice were injected with 7,8-DHF at 5 mg/kg i.p.; they were sacrificed 1 hour or 2 hours later, and amygdala tissue was collected bilaterally. Immunoblot analysis was performed with anti-TrkB Y706 (phosphorylated TrkB; Santa Cruz Biotechnology, Santa Cruz, Calif.; 1:200 in 5% bovine serum albumin/phosphate-buffered saline), and anti-TrkB (Cell Signaling Technology, Danvers, Mass.; 1:500 in 5% bovine serum albumin/phosphate-buffered saline), anti-
p-mitogen-activated protein kinase (MAPK; Cell Signaling Technology; 1:500 in 5% milk/phosphate-buffered saline), and anti-MAPK (Cell Signaling Technology; 1:5,000 in 5% milk/phosphate-buffered saline). For immunohistochemistry, fixed rat brain sections from prior studies (
11) were incubated and blocked in phosphate-buffered saline, goat serum, and Triton X-100 and incubated with TrkB rabbit polyclonal antibody (1:500, SC-12, Santa Cruz Biotechnology) and an anti-rabbit biotinylated secondary antibody (1:500) for 2 hours. Avidin-biotin complexes were amplified using a standard Vectastain Elite ABC kit (Vector Laboratories, Burlingame, Calif.). For regional localization of 7,8-DHF binding to TrkB, 7,8-DHF binding was performed as previously described (
18).
Cue Fear Conditioning and Extinction
Fear conditioning was conducted in nonrestrictive acrylic cylinders (SR-LAB startle response system, San Diego Instruments) located in a ventilated, sound-attenuated chamber. The footshock unconditioned stimulus was delivered through a stainless steel grid floor. Shock reactivity was defined as the peak activity (measured with a piezoelectric accelerometer) that occurred during the 200 msec after the onset of the unconditioned stimulus. The tone-conditioned stimulus was generated by a Tektronix function generator audio oscillator and delivered through a high-frequency speaker (
9,
19).
Note that fear conditioning includes a longer intertrial interval (5 minutes) between each of five conditioned stimuli to maximize fear learning, as compared to a greater number of stimuli (ranging from 15 to 30) for fear expression and extinction protocols, which use shorter intertrial intervals of 30-90 seconds. On each of 2 days prior to training, mice were given a 10-minute startle chamber exposure session to habituate them to handling and context.
During cued fear conditioning, mice received five trials of a conditioned stimulus tone (30 seconds, 12 kHz, 70 dB) coterminating with an unconditioned stimulus footshock (500 msec, 0.5 mA in experiments 1 and 2; 500 ms, 1 mA in experiments 3 and 4) with an intertrial interval of 5 minutes. The expression of fear (in a different context from training) was assessed 24 hours after fear conditioning and consisted of 15 conditioned stimulus tone trials of 30 seconds each, with a 1.5-minute intertrial interval. For extinction testing in experiments 2 and 4, mice were given 30 conditioned stimulus tone trials with a 30-second intertrial interval. Fifteen conditioned stimuli were presented in experiment 3 in the extinction session. Stimulus presentation and data acquisition were controlled and digitized by, and stored in, an interfacing desktop computer using SR-LAB and analyzed with the FreezeView software program (Coulbourn Instruments, Whitehall, Pa.).
Immobilization Stress
Immobilization procedures were conducted in a room separate from fear training and testing apparatus. Each animal was immobilized by gently restraining its four limbs in a prone position to metal arms attached to a wooden board for 2 hours. All animals of the same cage received the same treatment—either immobilization or handling. After treatment, animals were returned to their home cage and remained undisturbed until fear training.
More methodological details are provided in the data supplement that accompanies the online edition of this article.
Discussion
We have demonstrated that 7,8-DHF, a systemic TrkB agonist, enhances emotional learning. We have also built on prior in vitro data demonstrating that 7,8-DHF is a novel specific agonist at the TrkB receptor and that it crosses the blood-brain barrier to activate TrkB receptors in the brain, even in the absence of endogenous BDNF (
16,
17). Our data show that 7,8-DHF enhances several well-defined emotional learning and memory paradigms that have previously been shown to be BDNF dependent.
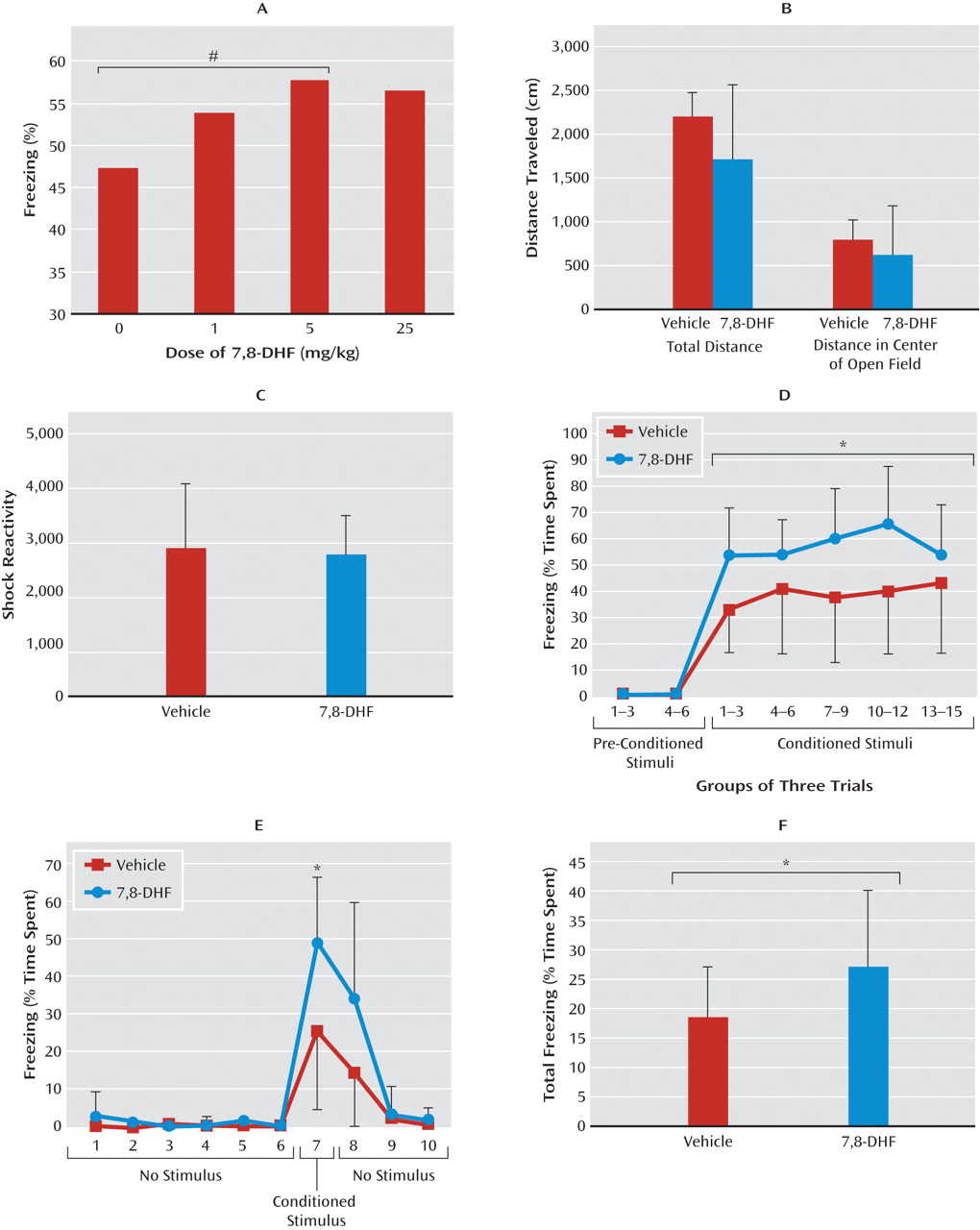
We first demonstrated that a single systemic dose of 7,8-DHF enhances the learning of cue-dependent fear conditioning in wild-type animals. This potentiated the fear response to the cue, suggesting that 7,8-DHF is likely targeting TrkB receptors in the brain that are normally critical for fear learning.
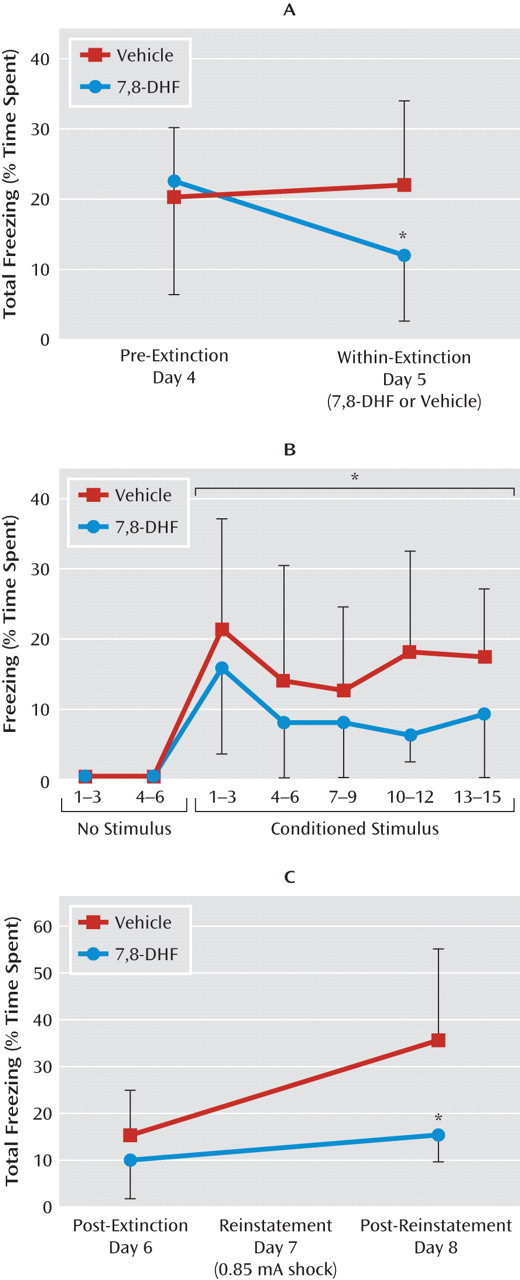
Extinction, or the specific new learning of fear inhibition, has also been shown to be BDNF dependent. We showed that 7,8-DHF could enhance extinction in wild-type mice. Clinical fear-related disorders have been shown to be related to a deficit in extinction, which may be related to the decreased BDNF found in a number of models of chronic stress. In our final experiment, we demonstrated that 7,8-DHF “rescued” a deficit in extinction of conditioned fear found in animals with a prior history of a single traumatic stress exposure.
Patients with PTSD and other anxiety disorders are thought to have deficits in extinction of aversive memories (
22,
23). Similarly, rodents with anxiety-like behavior or trauma exposure demonstrate a deficit in extinction of conditioned fear (
24–26). Notably, the finding that a single stressor is sufficient to impair later extinction is still quite novel and may have only one precedent in the literature (
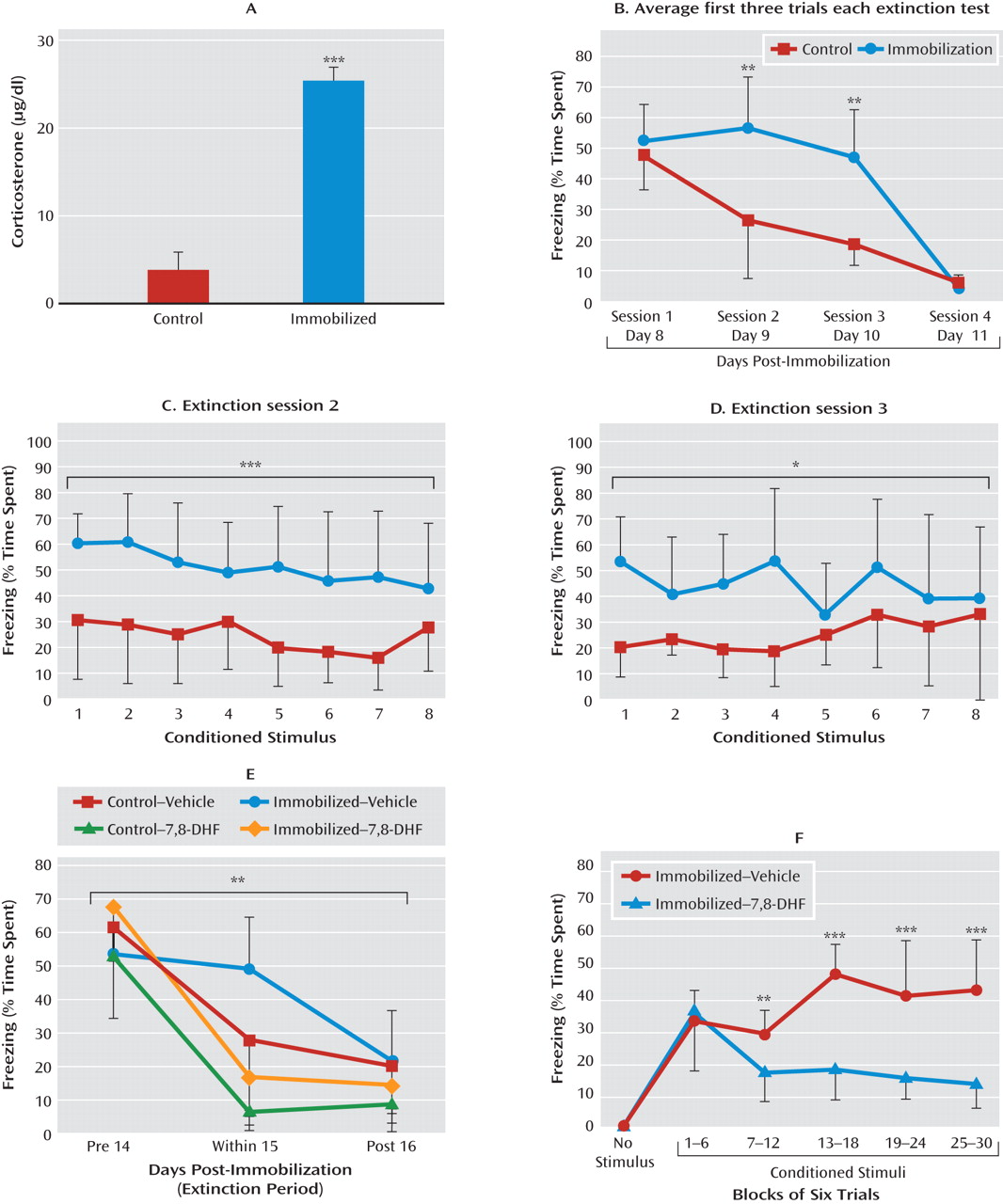
27). This disruption of extinction is thought to be due to both abnormalities in HPA-axis function and possible alterations in BDNF signaling. In mice, 2-hour immobilization led to a decrease in hippocampus BDNF protein levels at 5 and 10 hours after stress, but levels had returned to control levels when evaluated at 24 hours (
28). In rats, a single 2-hour period of stress immobilization has been found to be more stressful than high-intensity footshocks (
20). Similar stress has been shown to decrease BDNF mRNA in the hippocampus (
29), and immobilization has been shown to decrease BDNF mRNA levels in the amygdala and cortex (
30). In both cases, no difference was found in the TrkB receptor levels in any area. Together, these data suggest that even individual episodes of significant traumatic stress can alter BDNF function, which may affect later extinction processing.
Our data suggest that 7,8-DHF can fully “rescue” the deficit in extinction produced by prior immobilization stress. In fact, we found that 7,8-DHF exerts similar effects in both control and immobilization-stressed groups (
Figure 4E). This finding suggests that the TrkB receptor pathways may be intact after immobilization stress. Thus, the extinction deficit observed in previously immobilization-stressed mice may be due to defective release of BDNF or to a defect in factors other than the BDNF-TrkB pathway, but which can be rescued with additional activation of TrkB.
We found no effects on baseline behaviors or pain sensitivity following acute 7,8-DHF. These data suggest that 7,8-DHF did not have indirect effects that could alter the experience of fear training. Notably we found augmented learning for both fear and extinction, further supporting the role of the TrkB agonist in potentiating learning, and it was not due to enhancing or reducing sensory/motor function in a certain direction. Also, all of the learning paradigms performed here followed a single acute administration of systemic 7,8-DHF, which suggests that the emotional learning and memory enhancements were a function of direct and rapid activation of brain TrkB, as previously shown in vitro and in vivo (
16) and demonstrated for the first time here with autoradiography. Our data do not yet indicate where 7,8-DHF is acting in our fear acquisition and extinction studies. However, this study's immunoblots, autoradiography studies, and prior data from our group all suggest that TrkB in the amygdala, and possibly in the hippocampus and prefrontal cortex, could be involved in the systemic 7,8-DHF effects. Therefore, 7,8-DHF is likely augmenting emotional learning and plasticity, in the presence of endogenous BDNF, for both fear learning and extinction.
Overall, our findings suggest that this TrkB agonist may be an excellent research tool for understanding the effects of TrkB activation in a variety of learning and memory paradigms. Notably, the BDNF val/met polymorphism has been associated with increased anxiety-like behavior in humans (
31,
32) and mice (
33), as well as with altered extinction in both species (
34). Recently this mouse model was shown to have an extinction deficit that was reversed with
d-cycloserine (
35), an NMDA [
N-methyl-
d-aspartic acid]-dependent cognitive enhancer (
22). It will be fascinating to examine whether 7,8-DHF or similar TrkB-specific small-molecule agonists are able to reverse these effects in animal models and humans with the BDNF val/met polymorphism. Recent data also suggest that BDNF infusion into infralimbic cortex enhances extinction of fear (
36), and there is a burgeoning literature suggesting that decreased BDNF signaling may be a critical component in the pathophysiology of mood disorders (
15). Future research on the examination of systemically available BDNF agonists may lead to important, clinically relevant findings for the treatment of depression and anxiety disorders. Moreover, 7,8-DHF may be an attractive agent for improving extinction and other emotional learning deficits associated with psychopathology in humans.