A Trial of Prazosin for Combat Trauma PTSD With Nightmares in Active-Duty Soldiers Returned From Iraq and Afghanistan
Publication: American Journal of Psychiatry
Abstract
Objective
The authors conducted a 15-week randomized controlled trial of the alpha-1 adrenoreceptor antagonist prazosin for combat trauma nightmares, sleep quality, global function, and overall symptoms in active-duty soldiers with posttraumatic stress disorder (PTSD) returned from combat deployments to Iraq and Afghanistan.
Method
Sixty-seven soldiers were randomly assigned to treatment with prazosin or placebo for 15 weeks. Drug was titrated based on nightmare response over 6 weeks to a possible maximum dose of 5 mg midmorning and 20 mg at bedtime for men and 2 mg midmorning and 10 mg at bedtime for women. Mean achieved bedtime doses were 15.6 mg of prazosin (SD=6.0) and 18.8 mg of placebo (SD=3.3) for men and 7.0 mg of prazosin (SD=3.5) and 10.0 mg of placebo (SD=0.0) for women. Mean achieved midmorning doses were 4.0 mg of prazosin (SD=1.4) and 4.8 mg of placebo (SD=0.8) for men and 1.7 mg of prazosin (SD=0.5) and 2.0 mg of placebo (SD=0.0) mg for women. Primary outcome measures were the nightmare item of the Clinician-Administered PTSD Scale (CAPS), the Pittsburgh Sleep Quality Index, and the change item of the Clinical Global Impressions Scale anchored to functioning. Secondary outcome measures were the 17-item CAPS, the Hamilton Depression Rating Scale, the Patient Health Questionnaire–9, and the Quality of Life Index. Maintenance psychotropic medications and supportive psychotherapy were held constant.
Results
Prazosin was effective for trauma nightmares, sleep quality, global function, CAPS score, and the CAPS hyperarousal symptom cluster. Prazosin was well tolerated, and blood pressure changes did not differ between groups.
Conclusions
Prazosin is effective for combat-related PTSD with trauma nightmares in active-duty soldiers, and benefits are clinically meaningful. Substantial residual symptoms suggest that studies combining prazosin with effective psychotherapies might demonstrate further benefit.
Posttraumatic stress disorder (PTSD) is highly prevalent among active-duty U.S. military service members returning from combat deployments in Iraq and Afghanistan (1). Although a variety of psychotropic medications are used to treat PTSD symptoms in these service members (2), the medications’ efficacy and adverse effects in this population remain to be determined in controlled trials. In the absence of such trials, treatment guidelines for active-duty service members rely heavily on PTSD trials in civilian samples (3). Several issues characteristic of active-duty service members suggest caution in using this approach. These include the often high number and long duration of multiple combat-related traumatic stressors and the unavoidable reexposure to combat trauma reminders as personnel train for future combat deployments. It is also important to avoid sedation, weight gain, decreased libido, and other adverse effects of psychotropic drugs that can interfere with service members’ training, mission performance, and quality of life. Although the only two drugs approved for PTSD by the Food and Drug Administration (FDA) are selective serotonin reuptake inhibitors (SSRIs), it has been difficult to demonstrate SSRI efficacy for PTSD in U.S. military veterans (4–6).
Enhanced CNS adrenergic activity contributes to the pathophysiology of PTSD (7–9). Many PTSD symptoms, especially those assessed in the hyperarousal cluster of the Clinician-Administered PTSD Scale (CAPS) (10), are consistent with excessive CNS adrenergic activity (11). Prazosin is an inexpensive generic alpha-1 adrenoreceptor antagonist that reduces norepinephrine effects at CNS alpha-1 adrenoreceptors when administered peripherally (12). Prazosin does not produce sedation, sexual dysfunction, dyslipidemia, hyperglycemia, or weight gain. A daily bedtime dose of prazosin has been demonstrated to be effective for combat trauma PTSD nightmares, sleep disturbance, and global clinical status in Vietnam veterans (13, 14) and for sleep disturbance and total PTSD symptoms in younger veterans with low levels of combat trauma nightmares (15).
We hypothesized that prazosin would be an effective and well-tolerated treatment for combat trauma nightmares, sleep disturbance, and global function in active-duty soldiers with PTSD who have returned from deployments in Iraq or Afghanistan. Because prazosin has a short duration of action, study participants were given a midmorning and a bedtime dose to increase the likelihood of detecting prazosin efficacy for overall PTSD symptoms. We report the results of a prespecified interim analysis that prompted the Madigan Army Medical Center (MAMC) Institutional Review Board to discontinue enrollment because of demonstrated efficacy.
Method
The study was performed at MAMC, a large Army medical facility located on Joint Base Lewis-McChord (JBLM), Washington, and at VA Puget Sound Health Care System (VAPS). It was approved by JBLM command, the local MAMC and VAPS institutional review boards, and the Department of Defense Clinical Investigation Regulatory Office. Soldiers provided written informed consent after reading the consent form and having an opportunity to ask questions.
Participants
Active-duty soldiers (N=65) and recently discharged Army veterans (N=2) who had served in the Iraq or Afghanistan conflicts were recruited through banners and posters stating, “Health Studies: Sleep Disturbance With Combat Nightmares” with a contact telephone number self-referral. Soldiers also were referred by MAMC and JBLM health care providers who were made aware of the study through briefings (N=34) by the investigators (M.A.R., T.W., K.P., J.C., J.H., J.O.) and by fellow soldiers who had completed the study. Soldiers were enrolled between December 2009 and April 2011. Study visits were carried out at a JBLM medical clinic for active-duty soldiers and at VAPS for veterans.
Potential participants were screened by telephone for the presence of recalled distressing combat-related nightmares at least 2 nights per week. Those who met this major inclusion criterion were provided detailed information about the study, including the 50% chance of receiving placebo and the fact that open-label prazosin treatment was available. Those who elected to participate in the study were given an appointment for informed consent and a detailed eligibility evaluation by VAPS research personnel embedded 2 days a week at JBLM. Recruitment flow is presented in Figure S1 in the data supplement that accompanies the online edition of this article.
Inclusion and Exclusion Criteria
To be included in the study, participants had to meet DSM-IV criteria for PTSD; have a total score ≥50 on the 17-item CAPS; have a history of exposure to one or more life-threatening combat experiences that preceded onset of combat-related nightmares; have recalled distressing combat nightmares at least 2 nights/week; have a score ≥5 (maximum score of 8) on the CAPS nightmare item (item B2, “recurrent distressing dreams of the event”); and be willing to continue maintenance psychotropics (stable dosage for at least the 4 weeks prior to randomization) during the trial.
Medical exclusion criteria included acute or unstable medical illness; systolic blood pressure <110 mmHg supine or orthostatic hypotension (systolic blood pressure decrease from supine >20 mmHg after 2 minutes standing or any decrease accompanied by dizziness). Women were excluded if pregnant, currently nursing, or unwilling to use reliable birth control. Psychiatric and behavioral exclusion criteria, based on assessment with the Structured Clinical Interview for DSM-IV Axis I Disorders–Patient Edition (16), included psychotic disorders, cognitive disorders, substance abuse or dependence within the past 3 months, current cocaine or psychostimulant use, active suicidal or homicidal ideation, and depression requiring psychiatric hospitalization. Patients were also excluded if they were currently taking prazosin or any other alpha-1 adrenoreceptor antagonist or had a previous trial of prazosin for PTSD; if they had received prolonged exposure therapy, cognitive processing therapy, or eye movement desensitization and reprocessing therapy within 4 weeks before randomization; and if they had taken trazodone within 2 weeks of randomization (trazodone may increase the small risk of priapism associated with alpha-1 adrenoreceptor antagonists).
Randomization and Study Drug Titration
Eligible participants were assigned to receive prazosin capsules (1 mg, 2 mg, and 5 mg) or indistinguishable placebo capsules using a stratified permuted-block randomization procedure and stratified by current use of any antidepressant drug. On the drug initiation day, a clinician rater administered baseline evaluations. A separate clinician prescriber performed the dosage titration. The clinician raters and participants were blind to treatment condition. The clinician rater was also blind to the number of capsules/day prescribed, vital signs, and adverse events. Medication was titrated for up to 6 weeks with the goal of complete elimination of trauma nightmares. Medication was initiated at 1 mg at bedtime for 2 days and increased to 2 mg at bedtime for the next 5 days. The dosage was further increased at weekly intervals unless trauma nightmares were reported as absent during the preceding week, adverse effects were rated greater than mild, or the maximum allowed dosage had been reached. The maximum allowed dosage was 5 mg midmorning and 20 mg at bedtime for men and 2 mg midmorning and 10 mg at bedtime for women. The titration schedules are presented in Table 1. We set lower maximum morning and bedtime doses for women because of our and colleagues’ clinical observations of apparent increased sensitivity to both beneficial and adverse effects of prazosin in women with PTSD. The achieved daily dose of prazosin or placebo at the end of titration was continued as the maintenance dose for the duration of the 15-week trial.
| Men | Women | |||
|---|---|---|---|---|
| Week | Morning Dose (mg)a | Bedtime Dose (mg) | Morning Dose (mg)a | Bedtime Dose (mg) |
| Week 1 | ||||
| Days 1 and 2 | 1 | 1 | ||
| Days 3–7 | 2 | 2 | ||
| Week 2 | 1 | 4 | 1 | 2 |
| Week 3 | 2 | 6 | 1 | 4 |
| Week 4 | 2 | 10 | 2 | 6 |
| Week 5 | 5 | 15 | 2 | 10 |
| Week 6 | 5 | 20 | ||
a
The morning dose was taken between 10:00 and 11:00 a.m.
Assessments
The three prospectively designated primary outcome measures were the CAPS nightmare item, the Pittsburgh Sleep Quality Index (17), and the Clinical Global Impressions Scale (CGI) change item (18) operationalized as treatment impact on self-reported ability to function in daily activities. Secondary outcome measures were the 17-item CAPS; the three CAPS symptom clusters (reexperiencing, avoidance, and hyperarousal); the Hamilton Depression Rating Scale (HAM-D) (19); the Patient Health Questionnaire–9 (PHQ-9) (20); and the Quality of Life Inventory (21). Combat exposure was quantified with the Combat Experiences Scale (1), which was designed for deployment in Iraq and Afghanistan. Blood pressure after 5 minutes supine and after 2 minutes standing, along with any reported adverse events, were recorded at all visits. Behavioral ratings were obtained at baseline and at weeks 7, 11, and 15.
Statistical analyses for the primary, secondary, and exploratory outcome analyses followed the intent-to-treat principle. Differences between treatment groups in 15-week change from baseline were assessed using linear mixed-effects models, which include all participants and allow for missing values (22). These models included terms for gender, antidepressant use, week, treatment group, and, except in the case of the CGI change item, a week-by-treatment group interaction term; participants were treated as a random effect. Results for baseline and end of study are reported as adjusted means with the covariates gender and antidepressant use set to their average values. Results for differences between treatment groups in 15-week change from baseline are based on the week-by-treatment group interaction term.
Because the CGI change item inherently measures change from baseline, the linear mixed-effects model for this outcome measure did not include a week-by-treatment group interaction term and reported differences between treatment groups reflected the difference in CGI change item at week 15. Participants who dropped out before the week 7 visit did not have any CGI change item measures and were not included in the analyses of this measure.
As a secondary analysis, the 7-point CGI change item was collapsed into “responders,” defined as having markedly or moderately improved (scores of 1 or 2) and “nonresponders,” defined as minimally improved, no change, minimally worse, moderately worse, or markedly worse (scores of 3–7). The difference between treatment groups in the proportion of responders was assessed using a generalized linear-effects model with antidepressant use and gender as covariates.
An exploratory analysis addressed the possible effects of concurrent SSRI use (the predominant antidepressant class used) on change in the CAPS total score and the three primary outcome measures. For these analyses (except the CGI change item), the models included a week-by-treatment group-by SSRI use interaction term to determine whether the difference in change from baseline between treatment groups differed by SSRI use. The model for the CGI change item included a treatment group-by-SSRI use interaction term.
Adverse events were classified into 12 categories, as well as a category for any adverse event. The proportion of participants who experienced at least one adverse event in a category was compared between treatment groups using Fisher’s exact test. Confidence intervals for the differences between proportions were computed based on inverting the score statistic.
Results
A total of 67 participants underwent randomized treatment assignment; 35 were assigned to the placebo group and 32 to the prazosin group. Six participants in the placebo group (17%) and five in the prazosin group (16%) withdrew from the study before the first behavioral outcome rating at week 7 (see Figure S1 in the online data supplement). Of the 56 participants with at least one postrandomization behavioral rating (29 in the placebo group, 27 in the prazosin group), 46 completed the full 15 weeks (23 in each group), six completed 11 weeks (five in the placebo group, one in the prazosin group), and four completed 7 weeks (one in the placebo group, three in the prazosin group). Twenty participants were receiving SSRIs, and all had been maintained on the SSRI for at least 90 days. Of these 20 participants, 13 (65%) were assigned to the placebo group, of whom four (31%) dropped out before week 7, two (15%) completed 11 weeks, and seven (54%) completed all 15 weeks. Seven of the participants taking SSRIs (35%) were assigned to the prazosin group. Of these, one (14%) dropped out before week 7, one (14%) completed 7 weeks, and five (71%) completed all 15 weeks. The typical participant was a married male noncommissioned officer with substantial combat experience during two combat deployments (Table 2). Mean maintenance drug doses in men at both midmorning and bedtime were lower for the prazosin group than the placebo group (midmorning: 4.0 mg [SD=1.4] compared with 4.8 mg [SD=0.8]; bedtime: 15.6 mg [SD=6.0] compared with 18.8 mg [SD=3.3]; p values, <0.05). Achieved mean midmorning and bedtime doses in women did not differ significantly between the prazosin and placebo groups (midmorning: 1.7 mg [SD=0.5] compared with 2.0 mg [SD=0.0]; bedtime: 7.0 mg [SD=3.5] compared with 10.0 mg [SD=0.0]).
| Treatment Condition | ||||
|---|---|---|---|---|
| Characteristic | Prazosin (N=32) | Placebo (N=35) | ||
| Mean | SD | Mean | SD | |
| Age (years) | 30.0 | 6.6 | 30.8 | 6.5 |
| Educationa (years) | 13.3 | 1.9 | 13.0 | 2.1 |
| Combat Experiences Scale score | 10.9 | 3.8 | 11.9 | 3.6 |
| Number of deployments | 2.6 | 4.0 | 1.9 | 1.2 |
| N | % | N | % | |
| Male | 26 | 81 | 31 | 89 |
| Race/ethnicity | ||||
| African American | 4 | 13 | 5 | 14 |
| Asian | 1 | 3 | 0 | |
| Caucasian | 21 | 66 | 21 | 60 |
| Hispanic | 5 | 16 | 3 | 9 |
| Native American | 0 | 2 | 6 | |
| Other | 1 | 3 | 4 | 11 |
| Marital status | ||||
| Married | 19 | 59 | 26 | 74 |
| Widowed | 1 | 3 | 0 | |
| Separated/divorced | 8 | 25 | 5 | 14 |
| Never married | 4 | 13 | 4 | 11 |
| Major depression | 11 | 34 | 15 | 43 |
| Maintained on any antidepressant | 10 | 31b | 14 | 40c |
| Maintained on SSRI | 7 | 22 | 13 | 37 |
a
Missing values for three participants in each group.
b
All were on selective serotonin reuptake inhibitors (SSRIs) except one on amitriptyline, one on mirtazapine, and one on bupropion.
c
All were on SSRIs except one on mirtazapine.
Prazosin was significantly superior to placebo for all three primary outcome measures (Figure 1 and Table 3). The decrease in CAPS nightmare item score from baseline to endpoint was 3.1 (SE=0.3) in the prazosin group and 1.2 (SE=0.3) in the placebo group (difference in change from baseline, p<0.001; 95% CI=1.0–2.8). The decrease in score on the Pittsburgh Sleep Quality Index from baseline to endpoint was 5.6 (SE=0.7) in the prazosin group and 2.8 (SE=0.6) in the placebo group (difference in change from baseline, p=0.003; 95% CI=0.9–4.7). The proportion of CGI change item responders (markedly or moderately improved) was 64% (95% CI=44–79) for the prazosin group and 27% (95% CI=14–45) for the placebo group (difference in percent responders, p<0.001, odds ratio=4.8, 95% CI=1.9–12.2).
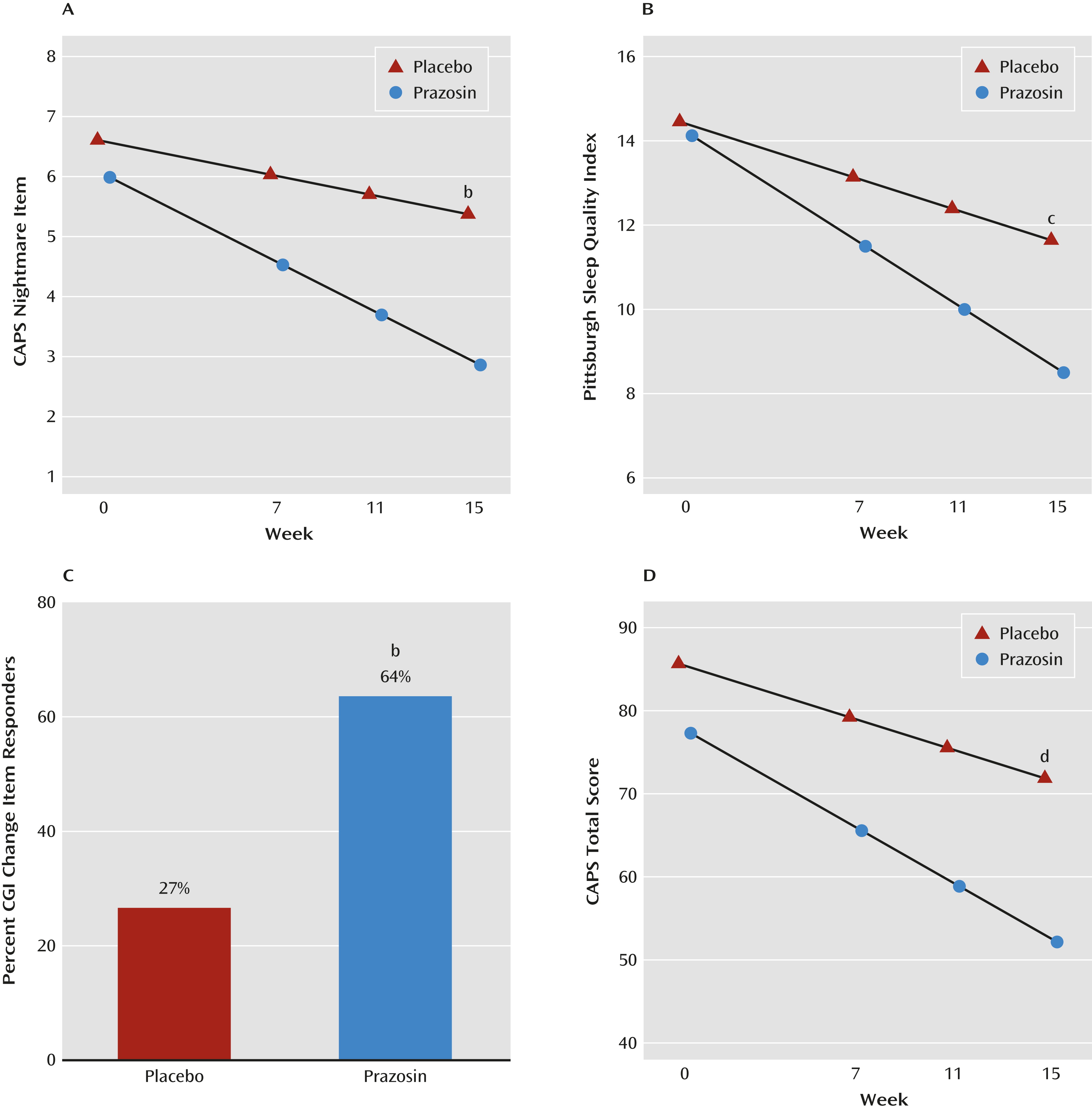
FIGURE 1. Effects of Prazosin on Symptom Ratings in Active-Duty Soldiers With Combat Trauma-Related PTSD With Nightmaresa
a Based on linear mixed-effects models. CAPS=Clinician-Administered PTSD Scale; CGI=Clinical Global Impressions Scale.
b Change from baseline to week 15 significantly greater for prazosin than placebo, p<0.001.
c Change from baseline to week 15 significantly greater for prazosin than placebo, p<0.01.
d Change from baseline to week 15 significantly greater for prazosin than placebo, p<0.05.
| Treatment Group | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Prazosin (N=32)b | Placebo (N=35)b | Difference in | ||||||||||
| Behavioral Outcome | Baseline | Week 15 | Baseline | Week 15 | Change From Baselinec | t | p | |||||
| Mean | 95% CI | Mean | 95% CI | Mean | 95% CI | Mean | 95% CI | Mean | 95% CI | |||
| CAPS nightmare item | 6.0 | 5.4, 6.6 | 2.9 | 2.2, 3.5 | 6.6 | 6.0, 7.2 | 5.4 | 4.8, 6.0 | 1.9 | 1.0, 2.8 | 4.06 | <0.001 |
| Pittsburgh Sleep Quality Index | 14.1 | 12.8, 15.5 | 8.5 | 7.1, 9.9 | 14.5 | 13.2, 15.7 | 11.6 | 10.3, 13.0 | 2.8 | 0.9, 4.7 | 2.98 | 0.003 |
| Clinical Global Impressions Scale change item | 2.3 | 1.8, 2.7 | 3.2 | 2.8, 3.6 | 0.9 | 0.3, 1.5 | 3.13 | 0.003 | ||||
| CAPS total score | 77.3 | 69.1, 85.5 | 52.2 | 43.8, 60.5 | 85.7 | 78.0, 93.3 | 71.9 | 63.9, 79.8 | 11.3 | 2.0, 20.7 | 2.39 | 0.02 |
| CAPS total score without nightmare item | 71.3 | 63.5, 79.1 | 49.3 | 41.3, 57.3 | 79.0 | 71.7, 86.4 | 66.5 | 58.9, 74.1 | 9.5 | 0.7, 18.3 | 2.12 | 0.04 |
| CAPS reexperiencing cluster | 22.3 | 19.8, 24.7 | 15.5 | 13.0, 18.0 | 25.1 | 22.7, 27.4 | 20.6 | 18.2, 23.1 | 2.3 | –1.2, 5.8 | 1.31 | 0.19 |
| CAPS avoidance cluster | 26.8 | 22.6, 31.0 | 18.2 | 13.9, 22.5 | 29.9 | 25.9, 33.8 | 24.7 | 20.6, 28.8 | 3.5 | –0.3, 7.3 | 1.81 | 0.07 |
| CAPS hyperarousal cluster | 28.2 | 25.7, 30.6 | 18.3 | 15.8, 20.8 | 30.7 | 28.4, 33.0 | 26.2 | 23.8, 28.7 | 5.4 | 1.9, 8.9 | 3.05 | 0.003 |
| Hamilton Depression Rating Scale | 11.9 | 9.5, 14.3 | 10.0 | 7.5, 12.5 | 14.7 | 12.4, 17.0 | 14.7 | 12.3, 17.1 | 2.0 | –0.8, 4.8 | 1.39 | 0.17 |
| Patient Health Questionnaire–9 | 12.1 | 9.9, 14.3 | 7.2 | 4.9, 9.4 | 14.7 | 12.6, 16.8 | 11.6 | 9.5, 13.8 | 1.8 | –0.5, 4.2 | 1.53 | 0.13 |
| Blood pressure (mmHg) | ||||||||||||
| Supine systolic | 126 | 123, 130 | 128 | 124, 131 | 126 | 123, 129 | 127 | 124, 130 | 0.1 | –4.1, 4.2 | 0.04 | 0.96 |
| Supine diastolic | 80 | 77, 83 | 81 | 78, 84 | 81 | 79, 84 | 82 | 79, 85 | –0.6 | –4.3, 3.2 | –0.29 | 0.78 |
| Standing systolic | 123 | 120, 126 | 124 | 120, 128 | 125 | 121, 128 | 124 | 121, 128 | –1.3 | –6.5, 3.8 | –0.51 | 0.61 |
| Standing diastolic | 82 | 79, 85 | 83 | 80, 86 | 84 | 81, 86 | 84 | 81, 87 | –0.9 | –4.8, 3.0 | –0.46 | 0.64 |
a
CAPS=Clinician-Administered PTSD Scale (17-item); CGI=Clinical Global Impressions Scale; Adjusted means and differences in 15-week change from baseline between treatment groups are based on linear mixed-effects models that include terms for gender and antidepressant use (these covariates are set to their average value). Change from baseline is defined as value at baseline minus value at week 15. For the CGI change item, change from baseline is value at week 15. For CGI change item responders (see text for results), adjusted percentages are based on a generalized linear mixed-effects model with covariates.
b
Total number of participants varies between time points and outcomes; at baseline, N=63–66; at week 15, N=48–50.
c
Difference in 15-week change from baseline between treatment groups (change from baseline in the prazosin group minus change from baseline in the placebo group). For the CGI change item, score at week 15 for the placebo group minus score at week 15 for prazosin group. For CGI change item responders (see text for results), the odds ratio for the prazosin group compared with the placebo group (based on generalized linear mixed-effects model with covariates).
The 17-item CAPS and the CAPS hyperarousal cluster demonstrated significantly greater improvement with prazosin than placebo (Figure 1 and Table 3). The mean change in total CAPS score from baseline to endpoint was 25.1 (SE=3.4) for the prazosin group, compared with 13.8 (SE=3.3) for the placebo group (difference in change from baseline, p=0.02; 95% CI=2.0–20.7). Total CAPS scores also were analyzed with the nightmare item removed. Differences favoring prazosin remained significant from baseline to end of study (mean=22.0 [SE=3.2], compared with mean=12.6 [SE=3.1]) (difference in change from baseline, p=0.04; 95% CI=0.7–18.3). The CAPS hyperarousal cluster improved significantly more with prazosin than placebo (p=0.003). Differences in the CAPS reexperiencing and avoidance clusters, the HAM-D, and the PHQ–9 depression scale numerically favored prazosin, but the differences did not reach statistical significance (Table 3). Using a 17-item CAPS score less than 20 as a criterion for full remission, three participants in the prazosin group and none in the placebo group achieved full remission.
For the total CAPS score outcome, there was a significant week-by-treatment group-by-SSRI use interaction (p=0.0007). Participants in the prazosin group not receiving an SSRI had a 15-week decrease from baseline of 30.1 points (SE=3.8), whereas those receiving an SSRI had a decrease of only 9.6 points (SE=6.8). Results were similar for the CAPS nightmare item (p=0.02; 15-week decrease in participants in the prazosin group not receiving an SSRI, mean=3.5 [SE=0.4]; in those receiving an SSRI, mean=1.9 [SE=0.7]) and the Pittsburgh Sleep Quality Index (p=0.01; 15-week decrease in participants in the prazosin group not receiving an SSRI, mean=6.4 [SE=0.8]; in those receiving an SSRI, mean=2.8 [SE=1.5]). The treatment group-by-SSRI use interaction for the CGI change item was not significant.
Two serious adverse events occurred during the study, both in participants in the placebo group. One participant was hospitalized for suicidal ideation. The other took a nonlethal overdose of oxycodone/acetaminophen as a suicide attempt. Other adverse events were generally mild and comparable between groups. Treatment-emergent adverse events associated with treatment for the prazosin group and placebo groups, respectively, were as follows: syncope, N=1 (3%) and N=0 (0%); lightheadedness, N=8 (25%) and N=7 (20%); nasal congestion, N=7 (22%) and N=4 (11%); lack of energy, N=0 and N=1 (3%); palpitations, N=2 (6%) and N=1 (3%); drowsiness, N=1 (3%) and N=3 (9%); depression, N=0 and N=2 (6%); and muscle weakness, N=1 (3%) and N=0. Miscellaneous adverse events occurred in 16 (50%) participants in the prazosin group and 23 (66%) in the placebo group. Headache was less frequent in the prazosin group (N=1 [3%]) than in the placebo group (N=8 [23%]); Fisher’s exact test, p=0.03). Blood pressure did not significantly differ over time or between treatment groups (Table 3). One brief syncopal episode occurred in a participant in the prazosin group on a maintenance dosage. It was judged likely to be related to prazosin in the context of dehydration during a physically demanding training exercise. The soldier continued in the study at full duty level and on his maintenance prazosin dosage with no further syncope or lightheadedness.
Discussion
This randomized controlled pharmacologic trial is, to our knowledge, the first reported for a behavioral disorder in active-duty U.S. combat service members. Prazosin was effective for all three primary outcome measures: combat-related trauma nightmares, sleep quality, and global status. Prazosin was also effective for overall PTSD symptoms even after the CAPS nightmare item was excluded. Previous research showed that in older Vietnam combat veterans with chronic PTSD, a single bedtime dose of prazosin was effective for trauma nightmares, sleep quality, and global status but not significantly superior to placebo for total CAPS score change (14). Because of its short duration of action (6 to 10 hours) (26), prazosin prescriptions in the medical management of hypertension or benign prostatic hypertrophy call for dosing two or three times daily. A midmorning dose in addition to a bedtime dose in the present study may have contributed to prazosin’s efficacy for overall PTSD symptoms.
The difference of 11.3 points in improvement in CAPS score between the prazosin and placebo groups exceeds the 9-point difference in CAPS score proposed as a threshold for clinical importance (27). This difference in CAPS score favoring prazosin is comparable to the differences favoring sertraline (6.8 points and 9.8 points) (28, 29) and paroxetine (10.8 points and 12.6 points) (30, 31) in the multicenter trials in predominantly civilian trauma samples that provided the databases for FDA approval of these SSRIs for the treatment of PTSD. Unexpectedly, greater PTSD symptom reduction with prazosin than with placebo was not reflected in scores on the Quality of Life Inventory. However, the clinical meaningfulness of prazosin’s effects on trauma nightmares, sleep quality, and total PTSD symptoms was supported by the high percentage of prazosin responders, as measured by global clinical status anchored to function.
The exploratory subgroup analysis of the effect of maintenance SSRI treatment on CAPS total score and the three primary outcome measures unexpectedly revealed a significantly smaller prazosin effect in participants taking SSRIs, except in the CGI change item. Because this was an unplanned post hoc analysis and the subgroup of participants in the prazosin group taking an SSRI was small (N=7), these results must be interpreted cautiously and may have occurred by chance. Also, because these participants (mean baseline CAPS score, 84 [SD=14]) had to have PTSD symptoms that were at least moderately severe to meet the study inclusion criterion of a total CAPS score ≥50, this criterion would select for individuals who were resistant to SSRI treatment and perhaps to other treatments as well. It is also possible that the modest adverse effects of SSRIs on sleep reduced the therapeutic effects of prazosin on trauma nightmares and sleep disturbance. Polysomnographic studies in healthy volunteers and patients with depression have demonstrated that SSRIs decrease sleep efficiency, total sleep time, and sleep continuity; increase light sleep; and decrease slow wave sleep (32). These effects on sleep are opposite to those of prazosin (14, 33). Clearly, prospective studies are needed to clarify hypotheses raised by these post hoc findings and to provide clinical guidance for using prazosin with an SSRI to treat PTSD in this population.
Prazosin was well tolerated at the high dosages achieved in these young adult soldiers. The sample size in this study is not sufficient to make definitive statements about the safety of prazosin, but adverse effects were no more frequent with prazosin than placebo despite continued participation in physically and mentally challenging training activities during the 15-week trial. The lower incidence of headaches in the prazosin group is consistent with a large open-label study in which prazosin markedly reduced headache frequency and severity along with sleep disturbance in Iraq veterans with mild traumatic brain injuries and a high prevalence of PTSD (34). The soldier who experienced syncope during intense physical exertion appeared to have been volume depleted. Although prazosin has been used safely to treat PTSD in soldiers participating in combat operations in the dehydrating Iraqi desert environment (35), maintaining adequate hydration during prazosin treatment remains important.
These results suggest that increased responsiveness to norepinephrine at the CNS alpha-1 adrenoreceptor contributes to the pathophysiology of PTSD. Excessive CNS noradrenergic activity is associated with irritability, sleep disturbance, and other hyperarousal symptoms typical of PTSD (11). Specific stimulation of CNS alpha-1 adrenoreceptors disrupts REM sleep (36), increases release of the anxiogenic neuropeptide corticotropin-releasing hormone (37), and favors “fight or flight” cognitive processes (38).
This study has several limitations. The sample was restricted to soldiers with frequent recalled combat trauma nightmares. Although we have demonstrated the efficacy of prazosin for trauma nightmares and overall PTSD symptoms in a small placebo-controlled crossover trial in a civilian PTSD sample (33), more studies in civilian trauma PTSD are needed. Also, results cannot be extrapolated to persons with PTSD who do not recall trauma nightmares. A retrospective chart review study suggested that prazosin might be useful for treating distressed awakenings in the absence of recalled trauma nightmares in veterans with chronic PTSD (39), but the efficacy of prazosin for PTSD-related distressed awakenings without recalled trauma nightmares remains to be prospectively studied. The present study could not evaluate whether improvement in PTSD symptoms persists after prazosin is discontinued because treatment responders elected to continue open-label prazosin.
Despite clinically meaningful effects of prazosin, the majority of soldiers continued to experience substantial PTSD symptoms. Treatment effect sizes and substantial residual PTSD symptoms of a magnitude similar to those in the present study were demonstrated in a large effectiveness trial of carefully supervised prolonged exposure therapy for PTSD in veterans (40). Studies combining prazosin with effective psychotherapies could result in further improvements in overall PTSD symptoms in active-duty service members and veterans.
Footnote
The views expressed are those of the authors and do not reflect the official policy of the Department of the Army, the Department of Defense, or the U.S. Government.
Supplementary Material
Supplementary Material (1003_ds001.pdf)
- View/Download
- 67.74 KB
References
1.
Thomas JL, Wilk JE, Riviere LA, McGurk D, Castro CA, Hoge CW: Prevalence of mental health problems and functional impairment among active component and National Guard soldiers 3 and 12 months following combat in Iraq. Arch Gen Psychiatry 2010; 67:614–623
2.
Morgan M, Lockwood A, Steinke D, Schleenbaker R, Botts S: Pharmacotherapy regimens among patients with posttraumatic stress disorder and mild traumatic brain injury. Psychiatr Serv 2012; 63:182–185
3.
Department of Veterans Affairs, Department of Defense: VA/DoD Clinical Practice Guideline: Management of Post-Traumatic Stress, version 2.0. Oct 2010 (http://www.healthquality.va.gov/PTSD-FULL-2010c.pdf)
4.
van der Kolk BA, Dreyfuss D, Michaels M, Shera D, Berkowitz R, Fisler R, Saxe G: Fluoxetine in posttraumatic stress disorder. J Clin Psychiatry 1994; 55:517–522
5.
Hertzberg MA, Feldman ME, Beckham JC, Kudler HS, Davidson JRT: Lack of efficacy for fluoxetine in PTSD: a placebo controlled trial in combat veterans. Ann Clin Psychiatry 2000; 12:101–105
6.
Friedman MJ, Marmar CR, Baker DG, Sikes CR, Farfel GM: Randomized, double-blind comparison of sertraline and placebo for posttraumatic stress disorder in a Department of Veterans Affairs setting. J Clin Psychiatry 2007; 68:711–720
7.
Southwick SM, Krystal JH, Morgan CA, Johnson D, Nagy LM, Nicolaou A, Heninger GR, Charney DS: Abnormal noradrenergic function in posttraumatic stress disorder. Arch Gen Psychiatry 1993; 50:266–274
8.
Mellman TA, Kumar A, Kulick-Bell R, Kumar M, Nolan B: Nocturnal/daytime urine noradrenergic measures and sleep in combat-related PTSD. Biol Psychiatry 1995; 38:174–179
9.
Geracioti TD, Baker DG, Ekhator NN, West SA, Hill KK, Bruce AB, Schmidt D, Rounds-Kugler B, Yehuda R, Keck PE, Kasckow JW: CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry 2001; 158:1227–1230
10.
Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Gusman FD, Charney DS, Keane TM: The development of a clinician-administered PTSD scale. J Trauma Stress 1995; 8:75–90
11.
Berridge CW: The locus ceruleus-noradrenergic system and stress: implications for post-traumatic stress disorder, in Post-Traumatic Stress Disorder: Basic Science and Clinical Practice. Edited by, Shiromani PJ, Keane TM, LeDoux JE. New York, Humana Press, 2009, pp 213–230
12.
Menkes DB, Baraban JM, Aghajanian GK: Prazosin selectively antagonizes neuronal responses mediated by alpha1-adrenoreceptors in brain. Naunyn Schmiedebergs Arch Pharmacol 1981; 317:273–275
13.
Raskind MA, Peskind ER, Kanter ED, Petrie EC, Radant A, Thompson CE, Dobie DJ, Hoff D, Rein RJ, Straits-Tröster K, Thomas RG, McFall MM: Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry 2003; 160:371–373
14.
Raskind MA, Peskind ER, Hoff DJ, Hart KL, Holmes HA, Warren D, Shofer J, O’Connell J, Taylor F, Gross C, Rohde K, McFall ME: A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry 2007; 61:928–934
15.
Germain A, Richardson R, Moul DE, Mammen O, Haas G, Forman SD, Rode N, Begley A, Nofzinger EA: Placebo-controlled comparison of prazosin and cognitive-behavioral treatments for sleep disturbances in US military veterans. J Psychosom Res 2012; 72:89–96
16.
First MB, Spitzer RL, Gibbon M, William JBW: Structured Clinical Interview for DSM-IV Axis I Disorders–Patient Edition (SCID-IP, Version 2.0). New York, Biometrics Research Department, New York State Psychiatric Institute, 1996
17.
Buysse DJ, Reynolds CF, Monk TH, Berman SR, Kupfer DJ: The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res 1989; 28:193–213
18.
Guy W: ECDEU Assessment Manual for Psychopharmacology: Publication ADM 76-338. Washington, DC, US Department of Health, Education, and Welfare, 1976, pp 218–222
19.
Hamilton M: A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56–62
20.
Kroenke K, Spitzer RL, Williams JB: The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 2001; 16:606–613
21.
Frisch MB: Manual and Treatment Guide for the Quality of Life Inventory–QOLI. Minneapolis, MN, Pearson Assessments, 1994
22.
Pinheiro JC, Bates DM: Mixed-Effects Models in S and S-PLUS. New York, Springer, 2000
23.
R Development Core Team: R: A Language and Environment for Statistical Computing, version 2.11.1. Vienna, Austria, R Foundation for Statistical Computing, 2011 (http://www.R-project.org)
24.
Pinheiro J, Bates D, DebRoy S, Sarkar D, R Core Team: nlme: Linear and Nonlinear Mixed Effects Models, R package, version 3.1-104, 2012. http://cran.r-project.org/web/packages/nlme
25.
Bates D, Maechler M, Bolker B: lme4: Linear mixed-effects models using S4 classes, R package, version 0.999999-0, 2012. http://CRAN.R-project.org/package=lme4.
26.
Westfall TC, Westfall DP: Adrenergic agonists and antagonists, in Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 11th ed. Edited by, Brunton L, Lazo T, Barker K. New York, McGraw-Hill, 2006, pp 237–296
27.
Krystal JH, Rosenheck RA, Cramer JA, Vessicchio JC, Jones KM, Vertrees JE, Horney RA, Huang GD, Stock C; Veterans Affairs Cooperative Study No. 504 Group: Adjunctive risperidone treatment for antidepressant-resistant symptoms of chronic military service-related PTSD: a randomized trial. JAMA 2011; 306:493–502
28.
Brady K, Pearlstein T, Asnis GM, Baker D, Rothbaum B, Sikes CR, Farfel GM: Efficacy and safety of sertraline treatment of posttraumatic stress disorder: a randomized controlled trial. JAMA 2000; 283:1837–1844
29.
Davidson JRT, Rothbaum BO, van der Kolk BA, Sikes CR, Farfel GM: Multicenter, double-blind comparison of sertraline and placebo in the treatment of posttraumatic stress disorder. Arch Gen Psychiatry 2001; 58:485–492
30.
Tucker P, Zaninelli R, Yehuda R, Ruggiero L, Dillingham K, Pitts CD: Paroxetine in the treatment of chronic posttraumatic stress disorder: results of a placebo-controlled, flexible-dosage trial. J Clin Psychiatry 2001; 62:860–868
31.
Marshall RD, Beebe KL, Oldham M, Zaninelli R: Efficacy and safety of paroxetine treatment for chronic PTSD: a fixed-dose, placebo-controlled study. Am J Psychiatry 2001; 158:1982–1988
32.
Oberndorfer S, Saletu-Zyhlarz G, Saletu B: Effects of selective serotonin reuptake inhibitors on objective and subjective sleep quality. Neuropsychobiology 2000; 42:69–81
33.
Taylor FB, Martin P, Thompson C, Williams J, Mellman TA, Gross C, Peskind ER, Raskind MA: Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma postraumatic stress disorder: a placebo-controlled study. Biol Psychiatry 2008; 63:629–632
34.
Ruff RL, Ruff SS, Wang XF: Improving sleep: initial headache treatment in OIF/OEF veterans with blast-induced mild traumatic brain injury. J Rehabil Res Dev 2009; 46:1071–1084
35.
Calohan J, Peterson K, Peskind ER, Raskind MA: Prazosin treatment of trauma nightmares and sleep disturbance in soldiers deployed in Iraq. J Trauma Stress 2010; 23:645–648
36.
Mallick BN, Majumdar S, Faisal M, Yadav V, Madan V, Pal D: Role of norepinephrine in the regulation of rapid eye movement sleep. J Biosci 2002; 27:539–551
37.
Vythilingam M, Anderson GM, Owens MJ, Halaszynski TM, Bremner JD, Carpenter LL, Heninger GR, Nemeroff CB, Charney DS: Cerebrospinal fluid corticotropin-releasing hormone in healthy humans: effects of yohimbine and naloxone. J Clin Endocrinol Metab 2000; 85:4138–4145
38.
Birnbaum S, Gobeske KT, Auerbach J, Taylor JR, Arnsten AF: A role for norepinephrine in stress-induced cognitive deficits: alpha-1-adrenoceptor mediation in the prefrontal cortex. Biol Psychiatry 1999; 46:1266–1274
39.
Thompson CE, Taylor FB, McFall ME, Barnes RF, Raskind MA: Nonnightmare distressed awakenings in veterans with posttraumatic stress disorder: response to prazosin. J Trauma Stress 2008; 21:417–420
40.
Eftekhari A, Ruzek JI, Crowley JJ, Rosen CS; National Center for PTSD: Effectiveness of national implementation of prolonged exposure therapy in VA care. JAMA Psychiatry (in press)
Information & Authors
Information
Published In


History
Received: 29 August 2012
Revision received: 3 January 2013
Revision received: 8 March 2013
Accepted: 13 March 2013
Published online: 1 September 2013
Published in print: September 2013
Authors
Funding Information
Supported by the Department of Veterans Affairs, U.S. Army Medical Research and Materiel Command, Fort Detrick, Md.; and by NIH grant 1R01MH069867.
Dr. Raskind has been an advisory board member for Janssen Immunotherapy, Pfizer Laboratories, and Baxter Pharmaceuticals. Dr. Peterson has been on the speakers bureau for Otsuka and AstraZeneca. Dr. Peskind has been an advisory board member for Lilly Pharmaceuticals and Avanir and on the speakers bureau for Forest Laboratories and Novartis. The other authors report no financial relationships with commercial interests.
Supplementary Material
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBMedia
Figures
FIGURE 1. Effects of Prazosin on Symptom Ratings in Active-Duty Soldiers With Combat Trauma-Related PTSD With Nightmaresa
a Based on linear mixed-effects models. CAPS=Clinician-Administered PTSD Scale; CGI=Clinical Global Impressions Scale.
b Change from baseline to week 15 significantly greater for prazosin than placebo, p<0.001.
c Change from baseline to week 15 significantly greater for prazosin than placebo, p<0.01.
d Change from baseline to week 15 significantly greater for prazosin than placebo, p<0.05.
Other
Tables
References
References
1.
Thomas JL, Wilk JE, Riviere LA, McGurk D, Castro CA, Hoge CW: Prevalence of mental health problems and functional impairment among active component and National Guard soldiers 3 and 12 months following combat in Iraq. Arch Gen Psychiatry 2010; 67:614–623
2.
Morgan M, Lockwood A, Steinke D, Schleenbaker R, Botts S: Pharmacotherapy regimens among patients with posttraumatic stress disorder and mild traumatic brain injury. Psychiatr Serv 2012; 63:182–185
3.
Department of Veterans Affairs, Department of Defense: VA/DoD Clinical Practice Guideline: Management of Post-Traumatic Stress, version 2.0. Oct 2010 (http://www.healthquality.va.gov/PTSD-FULL-2010c.pdf)
4.
van der Kolk BA, Dreyfuss D, Michaels M, Shera D, Berkowitz R, Fisler R, Saxe G: Fluoxetine in posttraumatic stress disorder. J Clin Psychiatry 1994; 55:517–522
5.
Hertzberg MA, Feldman ME, Beckham JC, Kudler HS, Davidson JRT: Lack of efficacy for fluoxetine in PTSD: a placebo controlled trial in combat veterans. Ann Clin Psychiatry 2000; 12:101–105
6.
Friedman MJ, Marmar CR, Baker DG, Sikes CR, Farfel GM: Randomized, double-blind comparison of sertraline and placebo for posttraumatic stress disorder in a Department of Veterans Affairs setting. J Clin Psychiatry 2007; 68:711–720
7.
Southwick SM, Krystal JH, Morgan CA, Johnson D, Nagy LM, Nicolaou A, Heninger GR, Charney DS: Abnormal noradrenergic function in posttraumatic stress disorder. Arch Gen Psychiatry 1993; 50:266–274
8.
Mellman TA, Kumar A, Kulick-Bell R, Kumar M, Nolan B: Nocturnal/daytime urine noradrenergic measures and sleep in combat-related PTSD. Biol Psychiatry 1995; 38:174–179
9.
Geracioti TD, Baker DG, Ekhator NN, West SA, Hill KK, Bruce AB, Schmidt D, Rounds-Kugler B, Yehuda R, Keck PE, Kasckow JW: CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry 2001; 158:1227–1230
10.
Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Gusman FD, Charney DS, Keane TM: The development of a clinician-administered PTSD scale. J Trauma Stress 1995; 8:75–90
11.
Berridge CW: The locus ceruleus-noradrenergic system and stress: implications for post-traumatic stress disorder, in Post-Traumatic Stress Disorder: Basic Science and Clinical Practice. Edited by, Shiromani PJ, Keane TM, LeDoux JE. New York, Humana Press, 2009, pp 213–230
12.
Menkes DB, Baraban JM, Aghajanian GK: Prazosin selectively antagonizes neuronal responses mediated by alpha1-adrenoreceptors in brain. Naunyn Schmiedebergs Arch Pharmacol 1981; 317:273–275
13.
Raskind MA, Peskind ER, Kanter ED, Petrie EC, Radant A, Thompson CE, Dobie DJ, Hoff D, Rein RJ, Straits-Tröster K, Thomas RG, McFall MM: Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry 2003; 160:371–373
14.
Raskind MA, Peskind ER, Hoff DJ, Hart KL, Holmes HA, Warren D, Shofer J, O’Connell J, Taylor F, Gross C, Rohde K, McFall ME: A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry 2007; 61:928–934
15.
Germain A, Richardson R, Moul DE, Mammen O, Haas G, Forman SD, Rode N, Begley A, Nofzinger EA: Placebo-controlled comparison of prazosin and cognitive-behavioral treatments for sleep disturbances in US military veterans. J Psychosom Res 2012; 72:89–96
16.
First MB, Spitzer RL, Gibbon M, William JBW: Structured Clinical Interview for DSM-IV Axis I Disorders–Patient Edition (SCID-IP, Version 2.0). New York, Biometrics Research Department, New York State Psychiatric Institute, 1996
17.
Buysse DJ, Reynolds CF, Monk TH, Berman SR, Kupfer DJ: The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res 1989; 28:193–213
18.
Guy W: ECDEU Assessment Manual for Psychopharmacology: Publication ADM 76-338. Washington, DC, US Department of Health, Education, and Welfare, 1976, pp 218–222
19.
Hamilton M: A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56–62
20.
Kroenke K, Spitzer RL, Williams JB: The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 2001; 16:606–613
21.
Frisch MB: Manual and Treatment Guide for the Quality of Life Inventory–QOLI. Minneapolis, MN, Pearson Assessments, 1994
22.
Pinheiro JC, Bates DM: Mixed-Effects Models in S and S-PLUS. New York, Springer, 2000
23.
R Development Core Team: R: A Language and Environment for Statistical Computing, version 2.11.1. Vienna, Austria, R Foundation for Statistical Computing, 2011 (http://www.R-project.org)
24.
Pinheiro J, Bates D, DebRoy S, Sarkar D, R Core Team: nlme: Linear and Nonlinear Mixed Effects Models, R package, version 3.1-104, 2012. http://cran.r-project.org/web/packages/nlme
25.
Bates D, Maechler M, Bolker B: lme4: Linear mixed-effects models using S4 classes, R package, version 0.999999-0, 2012. http://CRAN.R-project.org/package=lme4.
26.
Westfall TC, Westfall DP: Adrenergic agonists and antagonists, in Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 11th ed. Edited by, Brunton L, Lazo T, Barker K. New York, McGraw-Hill, 2006, pp 237–296
27.
Krystal JH, Rosenheck RA, Cramer JA, Vessicchio JC, Jones KM, Vertrees JE, Horney RA, Huang GD, Stock C; Veterans Affairs Cooperative Study No. 504 Group: Adjunctive risperidone treatment for antidepressant-resistant symptoms of chronic military service-related PTSD: a randomized trial. JAMA 2011; 306:493–502
28.
Brady K, Pearlstein T, Asnis GM, Baker D, Rothbaum B, Sikes CR, Farfel GM: Efficacy and safety of sertraline treatment of posttraumatic stress disorder: a randomized controlled trial. JAMA 2000; 283:1837–1844
29.
Davidson JRT, Rothbaum BO, van der Kolk BA, Sikes CR, Farfel GM: Multicenter, double-blind comparison of sertraline and placebo in the treatment of posttraumatic stress disorder. Arch Gen Psychiatry 2001; 58:485–492
30.
Tucker P, Zaninelli R, Yehuda R, Ruggiero L, Dillingham K, Pitts CD: Paroxetine in the treatment of chronic posttraumatic stress disorder: results of a placebo-controlled, flexible-dosage trial. J Clin Psychiatry 2001; 62:860–868
31.
Marshall RD, Beebe KL, Oldham M, Zaninelli R: Efficacy and safety of paroxetine treatment for chronic PTSD: a fixed-dose, placebo-controlled study. Am J Psychiatry 2001; 158:1982–1988
32.
Oberndorfer S, Saletu-Zyhlarz G, Saletu B: Effects of selective serotonin reuptake inhibitors on objective and subjective sleep quality. Neuropsychobiology 2000; 42:69–81
33.
Taylor FB, Martin P, Thompson C, Williams J, Mellman TA, Gross C, Peskind ER, Raskind MA: Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma postraumatic stress disorder: a placebo-controlled study. Biol Psychiatry 2008; 63:629–632
34.
Ruff RL, Ruff SS, Wang XF: Improving sleep: initial headache treatment in OIF/OEF veterans with blast-induced mild traumatic brain injury. J Rehabil Res Dev 2009; 46:1071–1084
35.
Calohan J, Peterson K, Peskind ER, Raskind MA: Prazosin treatment of trauma nightmares and sleep disturbance in soldiers deployed in Iraq. J Trauma Stress 2010; 23:645–648
36.
Mallick BN, Majumdar S, Faisal M, Yadav V, Madan V, Pal D: Role of norepinephrine in the regulation of rapid eye movement sleep. J Biosci 2002; 27:539–551
37.
Vythilingam M, Anderson GM, Owens MJ, Halaszynski TM, Bremner JD, Carpenter LL, Heninger GR, Nemeroff CB, Charney DS: Cerebrospinal fluid corticotropin-releasing hormone in healthy humans: effects of yohimbine and naloxone. J Clin Endocrinol Metab 2000; 85:4138–4145
38.
Birnbaum S, Gobeske KT, Auerbach J, Taylor JR, Arnsten AF: A role for norepinephrine in stress-induced cognitive deficits: alpha-1-adrenoceptor mediation in the prefrontal cortex. Biol Psychiatry 1999; 46:1266–1274
39.
Thompson CE, Taylor FB, McFall ME, Barnes RF, Raskind MA: Nonnightmare distressed awakenings in veterans with posttraumatic stress disorder: response to prazosin. J Trauma Stress 2008; 21:417–420
40.
Eftekhari A, Ruzek JI, Crowley JJ, Rosen CS; National Center for PTSD: Effectiveness of national implementation of prolonged exposure therapy in VA care. JAMA Psychiatry (in press)