Social anxiety disorder is highly prevalent and debilitating (
1), with an estimated prevalence of about 18% (
2,
3). The disorder is characterized by marked fearfulness and anxiety in social or performance situations, frequently resulting in avoidance and significant disability. In addition to suffering with social anxiety disorder, afflicted individuals often develop comorbid depressive and substance use disorders (
1). Data suggest that social anxiety disorder is ∼20%−40% heritable, with environmental factors accounting for the remaining variability (
4). Although social anxiety disorder is commonly diagnosed during adolescence, a period during which teenagers attempt to adjust to social change, it can start before adolescence, and its antecedents often manifest early in life (
5,
6). Accumulating evidence suggests that a behaviorally inhibited or temperamentally anxious disposition during childhood can lead to the development of social anxiety disorder (
5,
7). This at-risk phenotype is characterized by heightened, but nonpathological, levels of anxiety and may constitute a prodromal phenotype for social anxiety disorder. Although this phenotype is moderately stable over the course of development, it does not consistently predict the development of social anxiety disorder, nor is it invariant. This suggests that early-life interventions targeting highly anxious children have the potential to prevent the development of full-blown social anxiety disorder and its common comorbidities.
In this review, we take a cross-species approach to examine the behavior, neural circuits, and molecular systems that underlie the risk of developing social anxiety disorder. We discuss the biological basis of temperamental anxiety using data from humans and rodents, but we focus on insights gleaned from our studies of nonhuman primates. Rhesus monkeys are ideal for studying mechanisms underlying human development because, given their relatively recent evolutionary divergence from humans, the organization and function of the neural systems relevant to human anxiety, including the amygdala and prefrontal cortex, are conserved (
8,
9). Rhesus monkeys and humans also share similar complex social environments that rely on parent-child bonding and peer relationships. These early relationships can both encourage and discourage the adaptive social and emotional learning that helps regulate anxiety and promote survival. Our aim is to understand how inborn and environmental influences converge on the specific biological systems that underlie extreme temperamental anxiety and the risks it confers. It is our hope that understanding the mechanisms modulating these biological substrates will guide the development of novel early-life behavioral and pharmacological interventions that will provide effective treatment for children at risk of suffering from social anxiety disorder and related disorders.
The earliest manifestations of adaptive anxiety-related responses to potential threat occur during infancy and childhood; during infancy, they are characterized by increased excitability, and later, by behavioral inhibition. Behavioral inhibition occurs in response to novelty and potential threat and is associated with autonomic and pituitary-adrenal activation. Childhood behavioral inhibition is thought to manifest during the second year of life, around the time that a child emerges from the normative stage of stranger anxiety and is developing the ability to behaviorally cope with threat (
10,
11). Extreme behavioral inhibition, which has received considerable attention from the pioneering work of Jerome Kagan (
12), often manifests as excessive shyness and extremely reserved and avoidant behavior in social situations.
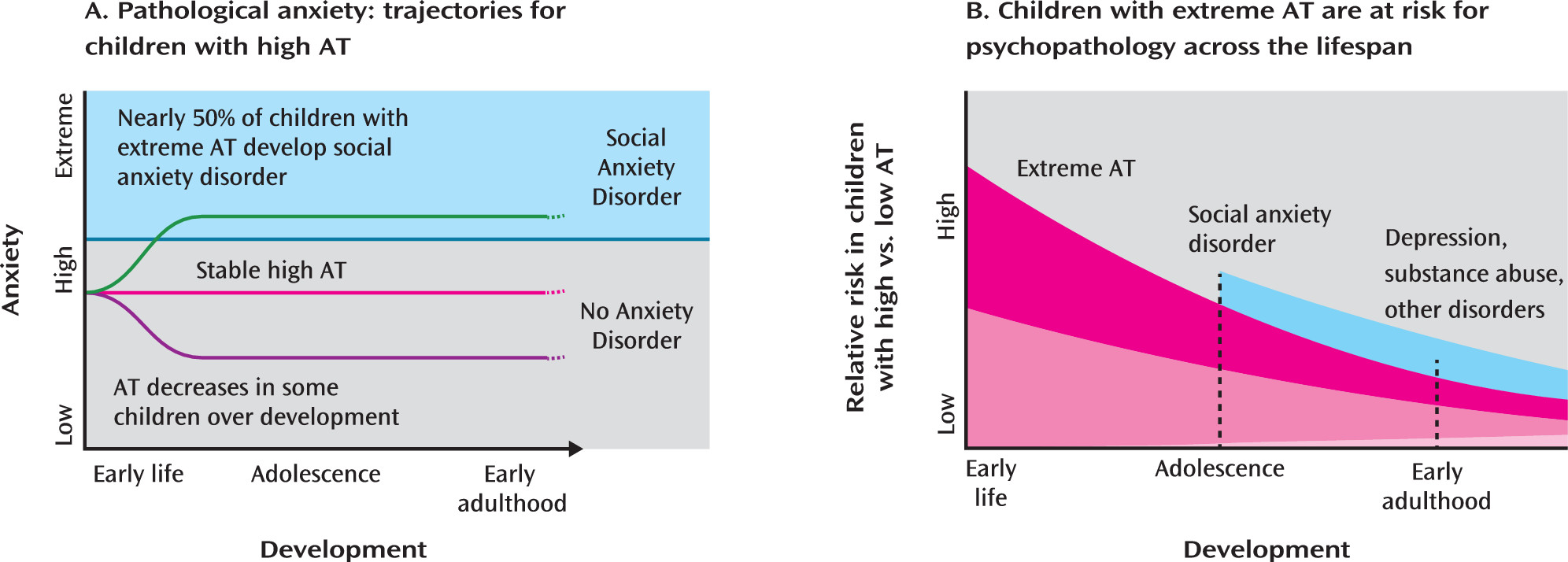
Several prospective longitudinal studies have found that extreme behavioral inhibition is associated with increased odds of developing anxiety and depressive disorders (
13–
18). A recent meta-analysis (
7) suggested that early-life behavioral inhibition is the single greatest predictor of the development of social anxiety disorder, as nearly 50% of highly behaviorally inhibited children go on to develop social anxiety disorder (
Figure 1A). Although behavioral inhibition fluctuates throughout childhood, studies suggest that individuals with stable levels of high behavioral inhibition have the greatest risk of developing social anxiety disorder (
15,
19–
21). Several prospective and retrospective self-report studies have implicated childhood behavioral inhibition as a risk factor for depressive disorders (
22–
24), and children of mothers with anxiety or depressive disorders tend to exhibit elevated levels of behavioral inhibition (
25–
27). Estimates of the heritability of behavioral inhibition are consistent with the estimates of ∼20%−40% for the heritability of anxiety disorders and anxiety-related neuroticism (
4,
28,
29). Many anxiety disorders, including those likely to develop later in highly behaviorally inhibited children, demonstrate partial shared heritability with each other (
4,
30,
31). These studies suggest that extreme behavioral inhibition is an early phenotype that confers risk for the development of a broad range of stress-related psychopathology (
32–
35) (
Figure 1B).
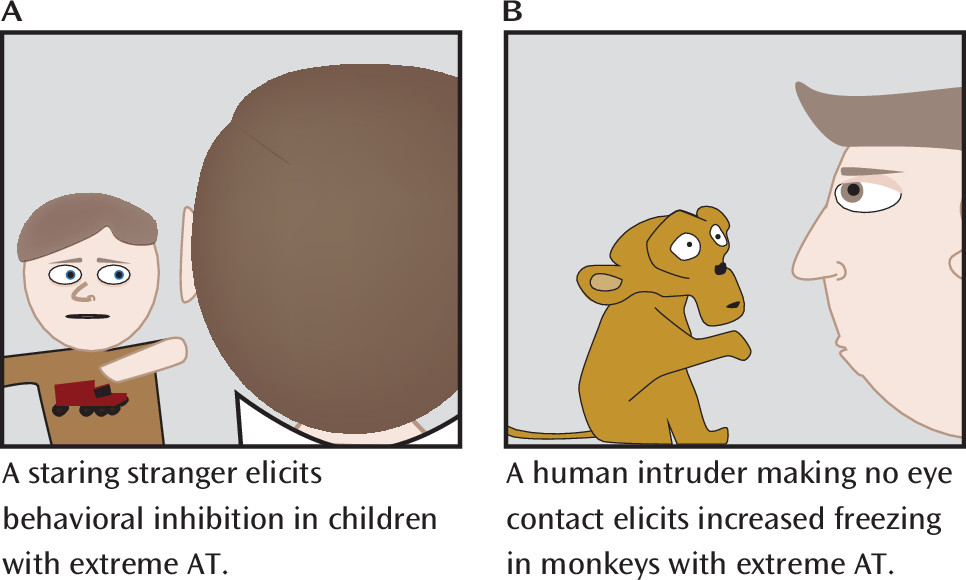
To further understand the biology of extreme early anxiety, our group has extensively validated a developmental rhesus monkey model. To assess behavioral inhibition, our initial studies developed the no-eye-contact (NEC) condition of the human intruder paradigm, in which the duration of freezing behavior, analogous to behavioral inhibition, was assessed in response to the uncertain threat of a human intruder looking away and presenting only his or her profile, being careful to make no eye contact with the monkey (
Figure 2) (
36). The NEC context can last for 30 minutes, during which the human intruder remains motionless, continuously presenting his or her profile throughout the entire test period. Our extensive studies examining various fear- and anxiety-related contexts have demonstrated that the NEC context specifically and reliably elicits freezing. In contrast, direct eye contact by the human intruder often elicits an overt aggressive response from the monkey. Freezing responses are evolutionarily conserved across diverse species, functioning to help the organism remain undetected in the presence of a potential predator (
37). Freezing is often accompanied by reduced coo vocalizations. Although rhesus monkey coo vocalizations are affiliative and can be used to recruit support from conspecifics, they can also attract predators. Thus, the reduction in coo calling during the NEC context is adaptive, as in the presence of a potential predator the value of remaining undetected is a survival imperative. The NEC context also induces physiological changes, such as increased cortisol levels and right-frontal EEG asymmetry (
38,
39). Moreover, similar to symptoms of anxiety in humans, behavioral inhibition in monkeys can be decreased with administration of the GABA-enhancing anxiolytic agent diazepam (
36).
To extend the assessment of behavioral inhibition, we developed the concept of anxious temperament (AT) to more completely reflect an individual’s dispositional physiological and behavioral responses to potential threat. In the monkey model, this composite AT measure is an average of standardized levels of NEC-context-induced freezing, coo vocalization reductions, and NEC-induced cortisol levels. More broadly, in relation to humans, we use the term AT to define the temperamental predisposition to display behavioral inhibition (increased freezing and decreased vocalizations) along with increased physiological reactivity (increased levels of the stress hormone cortisol) when exposed to novelty, unfamiliar individuals, or other potentially threatening situations. Compared with behavioral inhibition alone, we believe that the composite AT measure better estimates the at-risk human phenotype (
40).
Although there are no diagnostic criteria for mental disorders in nonhuman primates, data suggest that monkeys with high AT are functionally impaired. Anecdotally, the veterinary records from one extremely high AT monkey in our colony revealed significant stress-related symptoms, including hair loss, chronic diarrhea, and extreme fearfulness (e.g., refusal to take treats, retreating to the back of the cage, and excessive “crying”). We also characterized AT during the NEC context in a large free-ranging colony of monkeys on the island of Cayo Santiago. Naturalistic observations of these animals revealed that high AT, tested in a field-improvised laboratory, was associated with elevated social inhibition (unpublished data). Specifically, high AT predicted fewer conspecific approaches (r
s=−0.44, 0.007) when animals were free ranging, and high-AT animals maintained larger distances between themselves and their peers (r
s=−0.31, p=0.03). At the most extreme, some of the highest AT animals were never observed to approach their peers, while during the same period their low-AT counterparts approached their peers upwards of 50 times. These data add to the relevance of the extreme-AT monkey model as it relates to the dysfunction experienced by extremely anxious children. Further supporting the homology between human and monkey AT is our demonstration that individual differences in AT are relatively stable across development and that AT is ∼20%−40% heritable (
41,
42). Like human children, some high-AT monkeys exhibit a reduction in their AT levels as they mature, providing a unique opportunity for future studies to prospectively characterize brain mechanisms underlying recovery and resilience.
Brain Regions Associated With AT
In an initial functional MRI (fMRI) study, Schwartz et al. examined young adults who during their second year of life had been characterized as inhibited or uninhibited (
43). Strikingly, the results demonstrated that inhibited individuals had increased amygdala activation in response to novel neutral faces approximately 20 years after the original assessment. More recently, researchers demonstrated increased novelty-related amygdala activation in inhibited males approximately 18 years after being characterized as highly reactive at 4 months of age (
44). Similarly, young adolescents (∼12.5 years old) who were inhibited during childhood had increased amygdala activation when instructed to rate their emotional responses to fearful faces (
45). Extending these studies in prospectively characterized children, Blackford et al. (
46–
51) studied young adults who self-reported current and past behavioral inhibition. These studies provide additional evidence that amygdala activation is associated with an inhibited temperament, and they extend previous findings by implicating specific processes within the amygdala. Specifically, Blackford et al. demonstrated that highly inhibited young adults show faster amygdala response (
46), prolonged and exaggerated amygdala reactivity (
47,
48), decreased amygdala habituation (
49), and increased amygdala volume (
50). In addition to elucidating the role of the amygdala, Blackford et al. have begun to extend the set of regions associated with behavioral inhibition to include the hippocampus (
49), the lateral and medial orbitofrontal cortex (
47,
48), and the insular cortex (
48), as well as altered behavioral inhibition-related connectivity between these regions and the amygdala (
51).
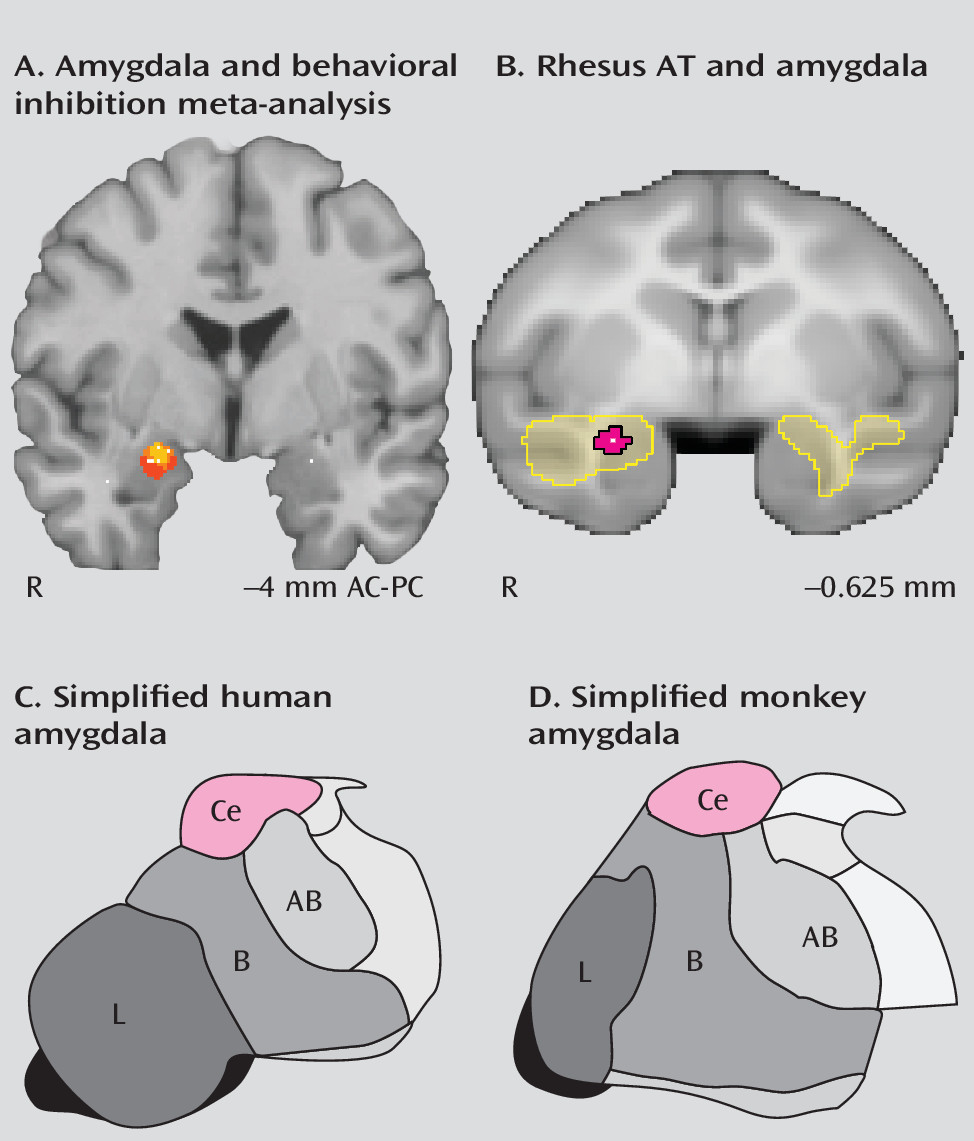
In neuroimaging studies of behavioral inhibition, the amygdala regions associated with behavioral inhibition are in the dorsal amygdala (
Figure 3A). The dorsal amygdala is anatomically distinct from the more ventrally located quasi-cortical basal and lateral amygdalar nuclei. The central nucleus of the amygdala, which is located within the dorsal amygdala region, is primarily composed of striatal-like GABA-ergic neurons and is considered to be the primary output structure of the amygdala complex (
53). In imaging studies, the precise localization of dorsal amygdala activations should be considered tentative, given the relatively large spatial confidence intervals associated with fMRI, particularly in studies examining relatively small groups of individuals (i.e., less than 50). Nevertheless, these studies suggest that the central nucleus may be important for instantiating the increased emotional reactivity characteristic of high-AT individuals.
fMRI studies in healthy individuals experiencing fear and anxiety have consistently identified an underlying neural circuit that includes the amygdala, the hippocampus, the prefrontal cortex, and the insula. Moreover, adults with anxiety disorders (e.g., social and specific phobia) show increased activation in these same regions (
54). Interestingly, novel social stimuli, such as emotional faces and eye whites, are sufficient to activate the amygdala (
55). Consistent with the continuity between dispositional anxiety and anxiety disorders, children with generalized anxiety disorder also show increased amygdala activation (
56). In addition to activation in the amygdala, children with generalized anxiety disorder demonstrate increased activation in prefrontal cortical area 47 near the anterior limb of the insula and increased amygdala-mid-insula functional coupling (
56).
To further examine the temperamental nature of anxiety-related brain metabolism, we examined young rhesus monkeys (1–4 years old, corresponding approximately to ages 3–12 years in humans) by phenotyping them for AT and performing [18F]fluorodeoxyglucose positron emission tomography (FDG-PET). Because the FDG-PET human intruder paradigm allows for the simultaneous assessment of brain metabolism and AT, this model can provide insights into the neural substrates underlying AT. As previously described, AT was assessed by combining the NEC-context-induced decreased spontaneous “coo” vocalizations (thought to reflect “calls for help”), increased freezing (or behavioral inhibition), and increased stress-induced cortisol levels (
36,
40). Our initial studies demonstrated that the components of AT are associated with metabolism in the extended amygdala, including the central nucleus of the amygdala and the bed nucleus of the stria terminalis (
Figure 3B), as well as the hippocampus, the anterior temporal lobe, and the brainstem periaqueductal gray matter (
57,
58). The extended amygdala comprises the central nucleus of the amygdala, the bed nucleus of the stria terminalis, and other forebrain structures that play an important role in the initiation of fear and maintenance of anxiety. Moreover, we found that our composite AT measure predicted significantly more variance in amygdala metabolism than any of the components that comprise AT (
40,
59). Later studies examining FDG-PET in relation to AT during the NEC context in more than 200 young rhesus monkeys revealed that metabolism in anterior temporal lobe structures, including the central nucleus of the amygdala, the anterior hippocampus, and the anterior temporal pole, robustly predicted AT (
41). These findings are consistent with human research in highly behaviorally inhibited individuals and in patients with anxiety disorders in that they provide evidence for the involvement of anterior temporal systems in the at-risk phenotype.
The fMRI and FDG-PET studies discussed above have been limited to studying brain activity in potentially stressful contexts—in the MRI scanner (
60) and in the NEC context. To further elucidate the temperamental nature of brain metabolism, we extended these studies to examine brain activity during nonstressful conditions. Specifically, we performed FDG-PET scans on animals that were each exposed to two different stressful conditions (NEC and separation from cagemate into a test cage), and two different nonstressful conditions (alone in the home cage without cagemate and life as usual in the home cage with cagemate). Trait-like positive correlations between individual differences in AT and metabolism in the amygdala, hippocampus, anterior temporal pole, and periaqueductal gray matter were found in each condition regardless of the level of stress (
40). Additionally, we examined the stability of AT’s neural substrates across time by assessing AT and FDG-PET in 24 animals that were exposed to the NEC context three times over 6–18 months. The results demonstrated interindividual stability over time in brain metabolism within AT-related regions (
61). Additionally, the mean metabolism across the three observations predicted the individual’s mean AT (
61). These data indicate that context-independent and temporally stable neural substrates underlie the trait-like nature of AT. These findings provide insight into AT, as they suggest that the neural substrates of AT are present even when no behavioral manifestations are apparent.
While our definition of AT is fairly circumscribed, there remains substantial variability in how AT presents. This variability is similar to the symptom heterogeneity observed within anxiety and affective diagnostic categories. For example, some monkeys display substantial freezing behavior while maintaining average levels of cortisol and emitting a normative number of coo calls. In contrast, other monkeys have high levels of cortisol relative to their behavioral responses. Studies in rodents by the Blanchards and others examining threat responses have suggested that while the activation of different physiological and behavioral responses can adaptively work together, they may also have different adaptive functions (
37). This raises the intriguing possibility that animals expressing different anxiety response profiles have tendencies to activate common neural circuits that can, through their effects on specific neural substrates, bias physiology and behavior toward different adaptive responses. In examining the neural substrates underlying AT’s components, we found both common and specific brain regions that underlie the phenotype’s heterogeneity (
59). The common brain regions included the central nucleus of the amygdala and anterior hippocampal regions, in which metabolism was independently associated with variation in freezing, cooing, and cortisol levels. This finding suggests that regardless of their “symptomatic” presentation, individuals with high levels of AT have increased metabolism in these brain regions. We also identified regional metabolism that was specific to each component of AT. For example, metabolism in the mid-hippocampus was uniquely associated with cortisol levels, as compared with freezing or coo vocalizations. Together these findings demonstrate that AT has both common and presentation-specific neural substrates and highlight the opportunity for understanding neural substrates that cut across phenotypic heterogeneity.
To understand how different brain systems relate to the heritability of AT, we used our large sample of brain imaging data from a multigenerational family pedigree to perform whole-brain heritability analyses. This was the first study to examine the heritability of brain metabolism across the entire brain. We were surprised to find differential heritability within AT’s neural substrates. Our results demonstrated significant heritability of anterior hippocampal metabolism, but no significant heritability of amygdalar central nucleus metabolism (
41). These findings call into question the view that it is solely amygdala-altering genes that are responsible for the intergenerational transmission of anxiety. Rather, our findings suggest that AT-related genes are more likely to exert their influence by altering function in other components of the AT-related circuit. Moreover, the lack of heritability within the central nucleus of the amygdala implies that this region may be more likely to mediate the environmental influences known to modulate AT, such as parenting, behavior modeling, and exposure to stress.
Causal Brain Regions and AT
Studies have been performed in patients with varying degrees of amygdala damage (
62–
64). Consistent with functional imaging findings, a rare patient with bilateral damage to her entire amygdala was shown not to experience psychological discomfort in response to invasions of her personal space (
65), did not have normative distrust of strangers (
66,
67), and did not report normal fearfulness (
68)—all features associated with decreased AT. This work supports the role of an amygdala-centered network in adaptive fear and anxiety as well as in anxiety disorders.
Targeted lesion studies in nonhuman primates reveal a causal role for dorsal amygdala regions in AT. Specific amygdala lesions decrease one’s reticence to act in potentially threatening situations and alter stress-induced cortisol release (
69–
71). Amygdala lesions also decrease anxiety in novel social situations, consistent with its role in social anxiety (
72,
73). Our studies employing specific neurotoxic lesions of the central nucleus of the amygdala (
74) demonstrated decreased freezing behavior and increased spontaneous coo vocalizations, two core components of AT. Although the central nucleus lesions did not directly affect cortisol, they reduced plasma concentrations of ACTH and CSF concentrations of corticotropin-releasing hormone, the two key upstream mediators of cortisol release.
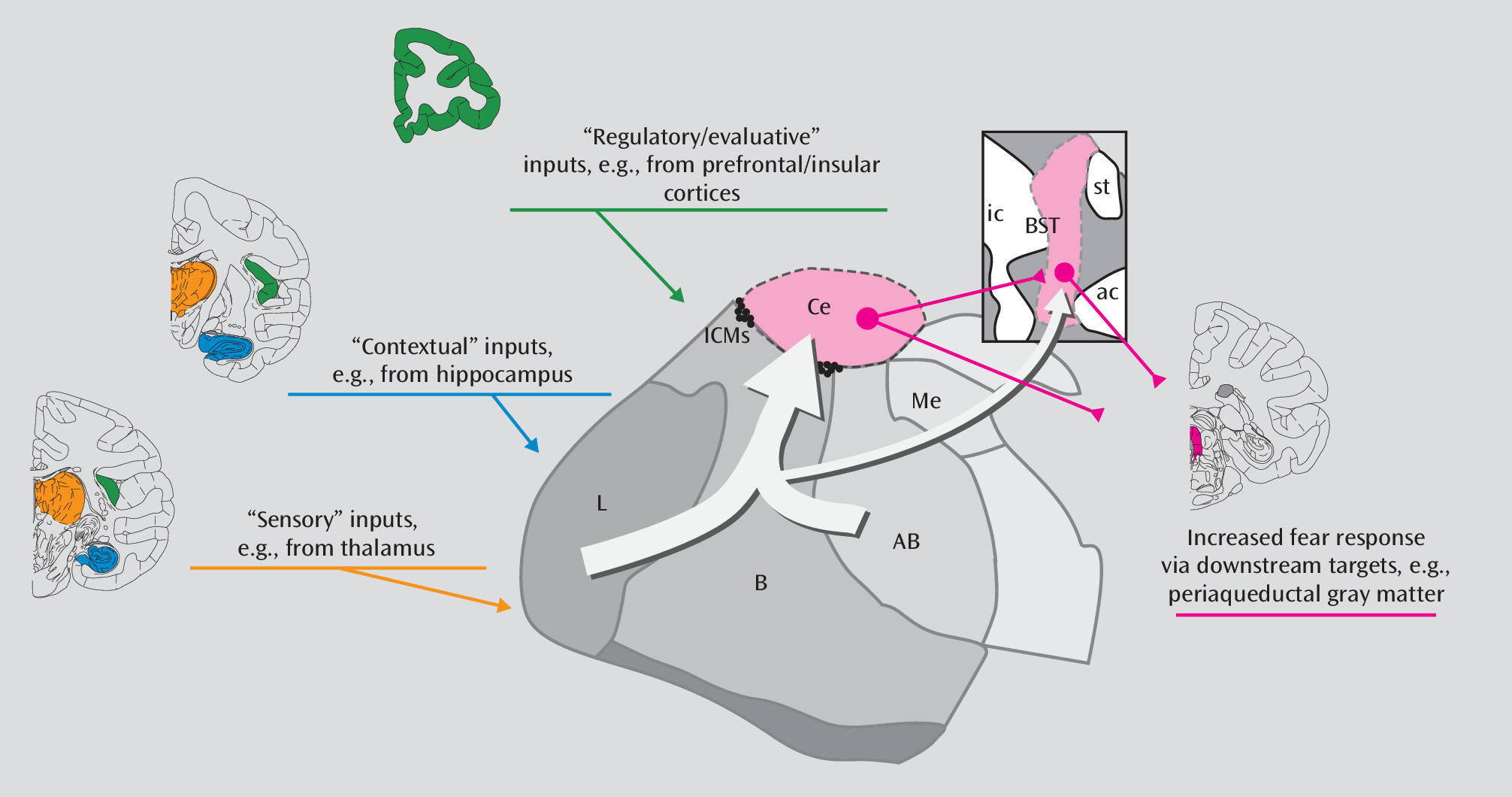
Targeted lesion studies in primates have also assessed the causal influences of the hippocampus and orbitofrontal cortex on components of AT. Both of these regions have direct connections to the amygdala and are thought to play regulatory roles and provide contextual/regulatory information to the amygdala (
Figure 4). Of particular interest is the finding that orbitofrontal cortex aspiration lesions decrease freezing behavior and cortisol levels (
70,
71,
75). By combining the lesion strategy with FDG-PET imaging, we found that the effects of orbitofrontal cortex lesions on AT could be explained by orbitofrontal cortex-induced changes in the extended amygdala (i.e., the bed nucleus of the stria terminalis), a region we previously found to be associated with AT (
76). Because orbitofrontal cortex aspiration lesions can also disrupt axons passing through this region, it is possible that the effects of these lesions are not due to orbitofrontal cortex damage per se, but rather result from damage to fibers originating in other prefrontal cortical regions. Consistent with this possibility, our recent fMRI study (
77) found that increased metabolism of the central nucleus of the amygdala and AT are associated with decreased central nucleus-dorsolateral prefrontal cortical intrinsic connectivity. In contrast to the effects of lesions of the orbitofrontal cortex and central nucleus, the evidence for hippocampal lesions affecting primate AT is mixed (
69,
70). Together, these data suggest that the central nucleus of the amygdala, the orbitofrontal cortex, and possibly the hippocampus may each causally influence AT, emphasizing the contribution of multiple regions to dispositional anxiety.
Lesion studies in rodents, although not assessing AT specifically, have demonstrated a causal role for many AT-related regions in unconditioned anxiety behaviors. In particular, rodent studies of unconditioned anxiety have causally implicated the amygdala (
78), the ventral hippocampus (similar to the anterior hippocampus in primates) (
79,
80), and the extended amygdala, including both the central nucleus of the amygdala and the bed nucleus of the stria terminalis (
78,
81). In both rodents and primates, the central nucleus of the amygdala and the bed nucleus of the stria terminalis project to the downstream structures necessary for initiating specific behavioral and physiological aspects of the fear response (
Figure 4). Elegant rodent studies have demonstrated dissociable roles for the central nucleus of the amygdala and the bed nucleus of the stria terminalis, such that the central nucleus is required for processing immediate and imminent threats, whereas the bed nucleus of the stria terminalis is required for responding to prolonged and more distant threats (
82). More recently, targeted optogenetic functional manipulations of specific projections and detailed anatomical studies have begun to elucidate projection- and cell-specific function within AT-related circuits. For example, some basolateral amygdala neurons provide excitatory input to the hippocampus and the central nucleus of the amygdala, which can initiate unconditioned fear- and anxiety-related behaviors (
83,
84). Moreover, specific subregions and cell types within the central nucleus of the amygdala and the bed nucleus of the stria terminalis have been demonstrated to mediate specific phenotypic expressions of anxiety (
85,
86). This suggests that selective alterations in the extended amygdala could give rise to the phenotypic heterogeneity observed in high-AT primates and humans with anxiety disorders. The rodent studies complement the human and nonhuman primate studies, strengthening support for the central nucleus of the amygdala in unconditioned anxiety and drawing attention to other components of the extended amygdala.
Molecular Processes Underlying AT
To develop ideal interventions aimed at preventing the long-term negative consequences of early-life AT, it is important to understand the molecular alterations occurring within the AT neural circuit. Genetic studies indicate that either extremely rare critical polymorphisms or many polymorphisms with small additive effects influence anxiety. Moreover, the influence of parents can have effects on anxiety via alterations in DNA methylation and other epigenetic phenomena that, in the case of methylation, can be passed down as alterations in parental methylation profiles and modified by parental behavior (
87–
89). Because mRNA levels reflect the confluence of genetic and environmental effects, we believe that examining gene expression within the neural substrates of AT can provide important clues. Because genetic methylation and expression profiles vary by tissue, region, and time, animal models are critical for developing a better understanding of the molecular alterations that underlie the altered brain function occurring in early-life AT.
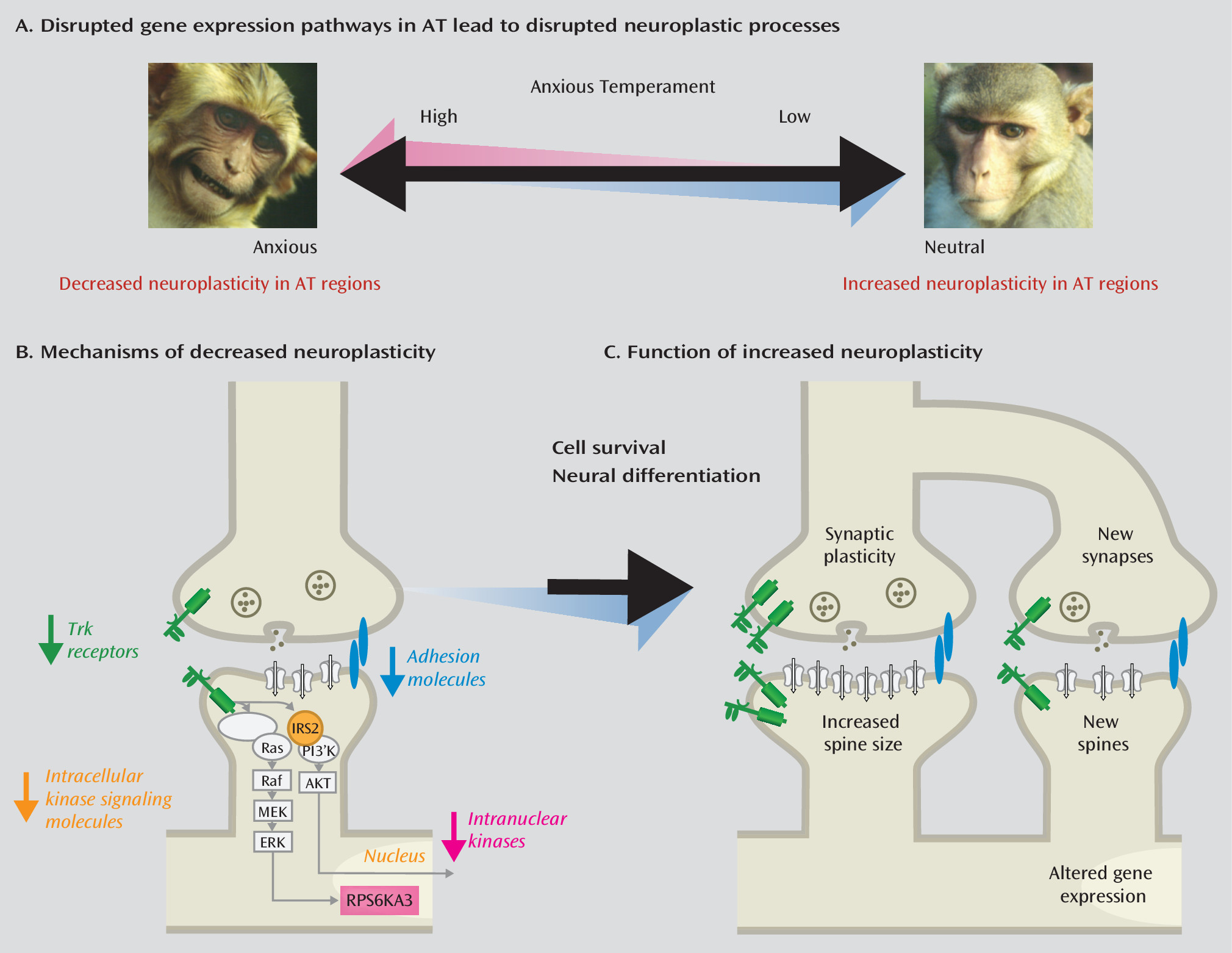
Because the strongest evidence linking brain alterations to AT points to the central nucleus of the amygdala, we examined individual differences in central nucleus gene expression in relation to AT (
61,
90). We performed prospective longitudinal brain imaging with behavioral and physiological assessments on 24 rhesus monkeys prior to measuring central nucleus gene expression. Altered gene expression occurred in some prominent anxiety-related neurochemical systems—neuropeptide Y and serotonin systems—such that individuals with high levels of
NPY1R or
5HT2C gene expression in the central nucleus demonstrated lower levels of AT. AT was also negatively associated with alterations in neurodevelopmental systems within neurotrophic and cellular adhesion pathways (
Figure 5). In particular, we observed a negative correlation between AT and the expression of neurotrophic receptor kinase 3 (
NTRK3, also known as
trkc) as well as its downstream partners, insulin receptor substrate 2 (
IRS2) and ribosomal protein S6 kinase, 90kDa, polypeptide 3 (
RPS6KA3, also known as
RSK2). These genes are involved in growth factor membrane signaling (
NTRK3), intracellular signaling (
IRS2), and nuclear activation (
RPS6KA3) (
Figure 5B), all of which contribute to synaptic plasticity and development (
Figure 5C). Importantly, individual differences in expression levels of
NTRK3 in the central nucleus of the amygdala predicted trait-like central nucleus metabolism.
NTRK3 is a growth-factor receptor that when activated can initiate widespread changes in cell growth and plasticity, similar to those seen after injection of
BDNF, which binds to
NTRK2 (also known as
TrkB). In relation to potential epigenetic mechanisms associated with AT, we observed that high-AT individuals had decreased levels of
GADD45B (growth arrest and DNA-damage-inducible, beta) in the central nucleus of the amygdala.
GADD45B is known to be involved in plasticity and neurogenesis through activity-dependent methylation of growth factors and thus may be relevant to the observed decreases in levels of
NTRK3 and its downstream partners (
91). Other nonhuman primate studies of early-life stress have also implicated neurodevelopmental pathways (
92,
93). Interestingly, recent research in squirrel monkeys demonstrated that coping in response to mild stress increases hippocampal neurogenesis and identified neurogenesis-related hippocampal gene expression in growth-related pathways that included
NTRK3 (
92).
Akil et al. have found similar results in relation to anxiety and depression (
94). Studies of gene expression in the frontal cortex of patients with major depressive disorder identified decreased expression of fibroblast growth factor 2 (
FGF2) (
95). Much like activation of NTRK3, activation of FGF2 can increase neurogenesis and synaptic plasticity.
FGF2 was also down-regulated in rodents bred to be behaviorally inhibited (
96). Excitingly, a single injection of FGF2 into the behaviorally inhibited rodents during the first days of life, prior to formation of the blood-brain barrier, was sufficient to decrease behavioral inhibition (
97). Follow-up study of these rodents revealed increased hippocampal neurogenesis and increased expression of neuroplasticity-related genes, including NTRK3, in relevant brain regions (
97). These data implicate the FGF family as important for the development of anxiety during early life and further support plasticity-related interventions aimed at decreasing AT (
94).
Researchers investigating the serotonin system in rodents have suggested that similar neuroplasticity-related mechanisms underlie anxiety and the efficacy of selective serotonin reuptake inhibitors (SSRIs). SSRIs are effective in treating anxiety disorders but often take weeks to fully work, suggesting an indirect mechanism. Stress impairs, whereas SSRIs increase hippocampal neurogenesis (
98,
99), and preventing hippocampal neurogenesis via irradiation blocks the effects of SSRIs (
99,
100). Recent studies have suggested that immature hippocampal neurons, in part, mediate the effects of SSRIs by enhancing the ability to discriminate complex threat-relevant information (
101,
102). Interestingly, the effects of SSRIs on neurogenesis seem to be mediated by the BDNF receptor, NTRK2 (i.e., TrkB). These data further support the role of tyrosine kinase pathways, as well as neuroplasticity, in relation to anxiety.
Research and Treatment Implications
Social anxiety disorder is common and debilitating. Lifelong social anxiety disorder often leads to further psychopathology, including mood and substance use disorders. Because it is possible to identify children at risk for social anxiety disorder early in life, the field has an unusual opportunity to conceptualize novel preventive intervention strategies. Treatments for social anxiety disorder are not completely effective, and no treatments exist for children with extreme AT, the forerunner of social anxiety disorder. While some children with extreme AT overcome their anxiety, early interventions promise to increase the number of children who grow up to be psychopathology free (
19,
21).
Evolutionarily conserved anxiety-related phenotypes have facilitated cross-species translational research. Studies of the neural circuits of social anxiety disorder and nonhuman primate AT implicate the dorsal amygdala, the anterior hippocampus, brainstem regions, and the orbitofrontal cortex. The homology between rhesus AT, childhood dispositional anxiety, and social anxiety disorder provides a framework for the valid use of nonhuman primates in new treatment development.
Alterations in brain function associated with AT are stable over time and are context independent. In contrast, the symptoms associated with AT and social anxiety disorder are elicited by specific cues and contexts associated with potential threat. These findings provide a conceptual basis for new treatments directed at changing the stable altered neural tendencies of individuals affected by AT and social anxiety disorder. Attempting to modify one’s trait-like brain function has the potential advantage of targeting mechanisms that may result in relapse and failure to respond. Lesions to different components of AT’s neural circuit diminish but fail to completely normalize AT. Thus, treatments targeting multiple AT-related brain regions are likely to be most successful in treating social anxiety disorder and preventing its development. Our research provides insights into which brain regions should be targeted. We identified anxiety-general regions that underlie anxiety regardless of how it is expressed and phenotype-specific regions that are uniquely involved in a particular expression of anxiety, such as freezing. Therefore, fully effective treatments will need to target anxiety-general regions as well as response-specific regions as they relate to the diverse presentations of individuals with AT and social anxiety disorder. The data demonstrate that the intergenerational transmission of anxiety is mediated by a widely distributed set of brain regions with large variation in the extent to which altered metabolism in these regions is heritable. This raises the possibility that optimal neural treatment targets could vary depending on one’s family history of anxiety. As treatments become more neuroscientifically focused, it is likely that neural measures reflecting treatment-related changes in AT’s neural circuits will be useful predictors of long-term treatment outcomes.
Characterizing the molecular alterations in brain regions associated with anxiety and AT has begun to identify novel treatment targets. Numerous rodent studies have focused on the hippocampus, where plasticity and neurogenesis have been associated with lower anxiety. Our group initially focused on the central nucleus of the amygdala for molecular analyses because the central nucleus is a core and causal component of AT’s stable neural substrate. Finding a reduction in neuroplasticity-related genes that are associated with increased central nucleus metabolism and AT led us to speculate that extreme early-life AT may result from a diminished ability to modify intra-central nucleus circuits. Studies of amygdala development in nonhuman primates have demonstrated that the central nucleus undergoes protracted development (
103), which seems to parallel the developmental course for children’s increased tendency to react by freezing to uncertainty and novelty (i.e., normative stranger anxiety). Thus, plasticity within this network is likely to be critical for the capacity to emerge from this period of heightened childhood fearfulness. We further hypothesize that the maturational ability to overcome or “unlearn” normative childhood fears relies on neuroplasticity mechanisms within AT’s neural substrates. Although the work described above specifically implicates
NTRK3,
FGF2, and other plasticity-related targets in relation to anxiety, we believe that the study results reflect involvement of broader neuroplasticity-related systems. Based on these results, it is likely that treatments that specifically increase neuroplasticity within the central nucleus and other components of AT’s neural substrates will be most effective in modulating early-life AT and preventing the development of social anxiety disorder. Neuroplasticity mechanisms in the hippocampus have been well studied and linked to antidepressant effects (
99,
100). Because of the central role of the central nucleus of the amygdala in AT, studies elucidating central nucleus-specific neuroplasticity molecular pathways will be important in conceptualizing novel treatments.
We have made the case for developing early interventions that are aimed at preventing high-AT children from developing anxiety disorders. Because AT emerges during a period when the brain is rapidly changing, treatments aimed at altering mechanisms underlying aberrant brain development are likely to have the potential for long-term changes in anxiety trajectories. Animal studies provide evidence for many ways to influence neuroplasticity that are relevant to treatment, such as exercise, SSRIs, and brain electrical stimulation. The building of synapses, neurons, and the resulting refinement of brain networks is a complex process involving many diverse molecules. Future work should focus on behavioral, pharmacological, and neuromodulatory strategies aimed at modulating diverse neuroplasticity-related molecules within specific components of AT’s neural substrates, such as the central nucleus of the amygdala and the anterior hippocampus.
In summary, research relevant to AT and the development of social anxiety disorder has revealed a number of important insights that can be helpful in formulating neuroscientifically based early-life interventions: 1) children with extreme AT are at high risk of developing further psychopathology, especially social anxiety disorder; 2) the neural circuits that underlie social anxiety disorder and human temperamental anxiety are similar to those implicated in monkeys with extreme AT; 3) the neural circuits that underlie AT are trait-like; 4) heterogeneous presentations of AT are associated with activity in both shared and phenotype-specific neural substrates; 5) environment and heritability differentially influence components of AT’s neural circuit; and 6) preliminary evidence points to altered neuroplasticity-related gene expression in the genesis of AT. It is our hope that this review will help focus research efforts on early interventions that are designed to not only reduce the suffering of anxious children but also to prevent them from developing further psychopathology.
Acknowledgments
The authors thank Jonathan A. Oler, Alexander J. Shackman, Do P.M. Tromp, Richard J. Davidson, Brad Postle, Wen Li, and Rick Jenison for their comments on early versions of the manuscript. They thank Steven E. Shelton, Helen VanValkenberg, Marissa Riedel, and the staff at the Harlow Center for Biological Psychology, the HealthEmotions Research Institute, the Waisman Center, the Waisman Laboratory for Brain Imaging and Behavior, and the Wisconsin National Primate Center.