Alzheimer’s disease begins with a long “preclinical” phase defined by the accumulation of brain deposits of fibrillar amyloid and pathological tau, a process spanning more than a decade before the onset of mild cognitive impairment (MCI) (
1–
3). Increasingly, observational studies have implicated depression and other neuropsychiatric symptoms as predictors of Alzheimer’s disease progression during this long preclinical period. Cognitively normal older people with neuropsychiatric symptoms, particularly depression-related symptoms and anxiety, have been found in multiple epidemiological cohorts (
3–
6) to be twice as likely to develop amnestic MCI, a prodromal phase of Alzheimer’s dementia, compared with those without these symptoms, over 3–6 years.
A small number of studies have investigated the relationship of in vivo markers of amyloidosis to syndromal depression or continuous measures of depression-related symptoms using cerebrospinal fluid (CSF) Alzheimer’s biomarker analyses and positron emission tomography (PET) imaging modalities in samples of cognitively normal older people. In cross-sectional analyses, Pomara and colleagues (
8) found that CSF amyloid beta 42 levels were reduced, as in Alzheimer’s disease, in cognitively intact elderly persons with late-life depression but not in those without depression, and that lower levels of amyloid beta 42 were inversely related to Hamilton Depression Rating Scale scores across the whole sample. CSF total tau and phosphorylated tau levels did not differ between the depressed and nondepressed groups. More recently, Babulal and colleagues (
9) found no cross-sectional association of CSF Alzheimer’s biomarkers with continuous scores of mood disturbance as assessed by the Profile of Mood States–Short Form (POMS-SF) in a cognitively normal community-dwelling sample. Notably, higher baseline CSF tau–amyloid beta 42 ratio, but not other CSF markers, predicted 1-year increases in anxiety and total mood disturbance scores on the POMS-SF and in total score on the Neuropsychiatric Inventory Questionnaire, suggesting a dynamic relationship between Alzheimer’s-specific markers and changes in emotional tone. In related findings, mean cortical binding of fibrillar amyloid determined by Pittsburgh compound B (PiB) PET imaging was not associated with depression or other neuropsychiatric symptom measures at baseline but was positively associated with change scores on the 15-item Geriatric Depression Scale (GDS-15) over 1 year. Similarly, Harrington and colleagues (
10), using PET radioligands to classify a large, nondepressed, cognitively normal sample into high and low amyloid beta groups, found that the high amyloid beta group was 4.5 times more likely than the low amyloid beta group to develop high categorical depression, defined by the GDS-15, after 54 months, in unadjusted analyses.
Together these recent findings suggest that cognitively normal older individuals with biomarker evidence of amyloidosis (principally high fibrillar brain amyloid beta levels) are more likely to experience rising depressive symptoms over time. At the same time, these preliminary observations raise more pointed questions as to the quality, severity, and time course of depressive symptoms that are most characteristic of preclinical Alzheimer’s disease and the specificity of their associations with Alzheimer’s molecular markers. To approach these questions, we investigated the relationship of brain amyloid beta burden, determined by PiB-PET, to longitudinal measures of depression in a cohort of cognitively normal older adults comprising individuals with a wide range of amyloid beta values, including a subset with a high amyloid beta burden consistent with preclinical Alzheimer’s disease. We hypothesized that higher amyloid beta levels would predict greater depression scores, even in a subclinical range, after adjustment for other potential confounders, including depression history. Building on previous work (
11) that defined latent factors of subclinical depressive symptoms within the same cohort, we also examined associations of amyloid beta and depressive symptom clusters over time.
Method
Participants
Data were derived from the Harvard Aging Brain Study (HABS), an observational study of older adult volunteers, aimed at defining neurobiological and clinical changes in early Alzheimer’s disease. A total of 270 participants completed study visits over 5 years (mean number of visits, 3.8; range, 1–5). Participants were English-speaking, community-dwelling men and women, ages 62–90, who were cognitively normal and had no active major psychiatric disorders at the time of enrollment (
11). A history of past or current depression adequately treated with standard antidepressant medication (selective serotonin reuptake inhibitors or dual serotonin-norepinephrine reuptake inhibitors, bupropion, mirtazapine, trazodone, or nortriptyline) was allowed. At screening, all participants scored below cutoff for mild depression, defined as a score ≥11 on the 30-item Geriatric Depression Scale (GDS) (
12). Cognitively normal status was defined by a Clinical Dementia Rating (
13) global score of 0 and education-adjusted normal performance on the logical memory subtest of the Wechsler Memory Scale (
14) and the Mini-Mental State Examination (MMSE) (
15). The Partners Human Research Committee approved the study, and all participants provided written informed consent.
Clinical Measures
Baseline clinical assessments relevant to these analyses included the MMSE; the American National Reading Test intelligence quotient (AMNART) (
16), a measure of premorbid intelligence; and the two-factor Hollingshead score, calculated according to primary occupation and educational attainment (range in the sample, 11–73; higher score indicates lower socioeconomic status) (
17). Participants were classified by genotype as apolipoprotein E ε4 (APOEε4) allele carriers or noncarriers. Self-reported depression and treatment were elicited at baseline via history obtained by a study physician, followed by medical record review if necessary. Depression history was defined as a dichotomous variable in which participants with any self-reported depression, including past or current diagnoses, were classified together and compared with participants with no depression history.
Depression was quantified at baseline and annually using the GDS (item range, 0–1; total range, 0–30; higher score indicates greater depression) (
12). In addition to calculating a total score for each time point, we also calculated an average score corresponding to each of three clusters of GDS items. These aggregate GDS items, the anxiety-concentration, apathy-anhedonia, and dysphoria clusters, were previously defined by principal component analysis using a HABS sample that was nearly identical to the baseline sample used in the present study (
11) (see the Methods section in the
data supplement that accompanies the online edition of this article).
All depression data were acquired in a blinded fashion with regard to other assessments and procedures.
PiB-PET Data
Fibrillar amyloid burden was measured using PiB-PET, according to established protocols, at the Massachusetts General Hospital PET facility (
18–
21). PiB distribution volume ratio (DVR) was calculated according to methods established in previous studies that include a single region representing the aggregate cortical areas at risk for amyloid burden, across the frontal, lateral temporal, and lateral and medial parietal lobes (
21,
22). Analyses used aggregate PiB DVR as a continuous measure. Participants and investigators were blind to all PiB data. PiB DVR data across the full range of values were analyzed.
Statistical Analysis
Unadjusted associations between baseline GDS total score, GDS cluster scores, and PiB measures were tested using Pearson correlations. Relations among categorical predictors were evaluated using chi-square tests, and differences in mean relations between categorical predictors and baseline numerical variables were assessed with Satterthwaite t tests.
Mixed random- and fixed-effects longitudinal analyses were run across time in the study for each of four dependent variables (GDS total score and average scores for the anxiety-concentration, dysphoria, and apathy-anhedonia clusters) in separate analyses, employing a backward elimination algorithm (p<0.05 cutoff) on an initial pool of fixed predictors and variances/covariances of random terms. During backward elimination, by convention, nonsignificant terms are retained in the model if higher-order terms subsuming them, e.g., interactions, are still in the model. The time predictor was the linear component of years in the study (preliminary graphs did not suggest curvilinearity). Fixed terms were baseline PiB DVR, age at baseline, history of depression (yes/no), AMNART score, Hollingshead score, sex, APOEε4 carrier status (yes/no), and the interaction of each of these predictors with time in study. Antidepressant use was not included as a predictor, as this substantially overlapped with depression history. Random terms were intercepts and linear slopes across time per participant, initially allowing for a correlation between them. Percent variance accounted for in the dependent variable by fixed and random predictors was computed.
SAS, version 9.4 (SAS, Cary, N.C.), SPSS, version 23 (IBM, Armonk, N.Y.), and R, version 3.3.2 were used.
Results
Demographic, clinical, and imaging data are summarized in
Table 1 and in Figure S1 in the
online data supplement. The mean GDS score at baseline was 2.8 for the whole sample and 3.7 for participants with a history of depression. Average scores for items corresponding to the apathy-anhedonia and the anxiety-concentration clusters were greater than for the dysphoria cluster (mean values, 0.161, 0.129, and 0.030, respectively, where these are equivalent to the mean proportion of cluster items that participants endorsed; see the Methods section in the
data supplement). At baseline, anxiety-concentration scores were significantly but weakly correlated with the other cluster scores (for apathy-anhedonia, r=0.2, p<0.0001; for dysphoria, r=0.2, p=0.0003), whereas the other clusters were not significantly correlated with each other (for apathy-anhedonia and dysphoria, r=0.1, p=0.09). At baseline, PiB binding was not significantly correlated with GDS total score (r=0.08, p=0.2), anxiety-concentration score (r=0.03, p=0.6), dysphoria score (r=−0.01, p=0.9), or apathy-anhedonia score (r=0.1, p=0.06).
Thirteen percent (N=35) of the participants reported a history of depression: 7% of participants reported active depression within 2 years of enrollment (current depression) and 6% reported past depression but no active depression within 2 years of enrollment (past depression). Participants with and without a history of depression did not differ proportionally across sex or APOEε4 categories (
Table 1). Participants with a history of depression, compared with those with no depression history, had a significantly higher mean baseline PiB binding (
Table 1). More specifically, a significant difference in mean PiB binding was found between those with no depression history compared with those with current depression (1.16 and 1.28, respectively; p=0.006) but not compared with those with past depression (1.16 and 1.22, respectively; p=0.2).
Longitudinal Analyses
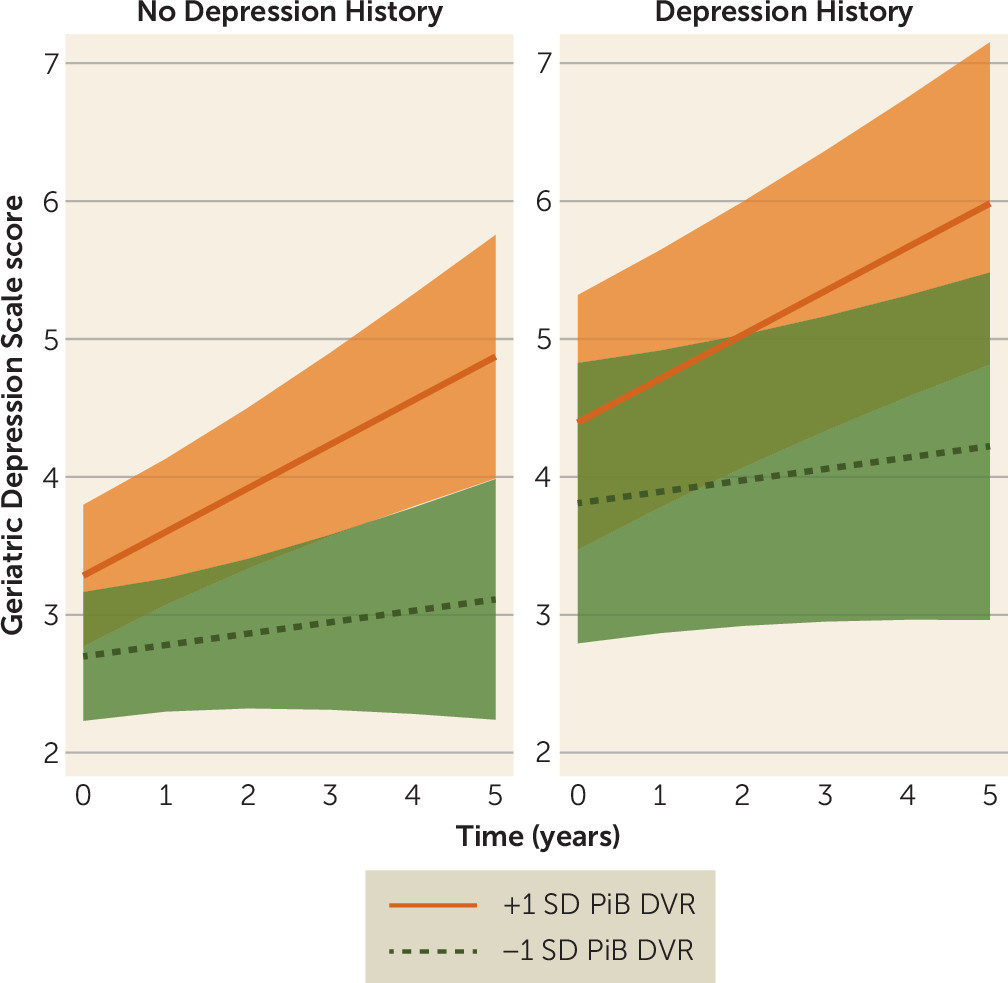
In the final model for GDS score, participants with a history of depression had a higher adjusted mean GDS score across time compared with those without self-reported depression (
Table 2). The interaction of PiB binding with time was also a significant predictor in this model, such that higher baseline PiB binding associated with steeper increases in GDS score over time (
Figure 1). Neither APOEε4 nor its interaction with time was associated with higher GDS score, and no other fixed terms were significant. Significant random terms were an uncorrelated intercept and linear slope across time. In a post hoc model, we repeated the final model, testing for possible effect modification based on APOEε4 status. Terms for the multiplicative interactions of PiB binding by APOEε4 status and PiB binding by APOEε4 status by time were added as predictors to the final model and were not significantly associated with GDS score.
As in the model for GDS total score, final predictors of anxiety-concentration scores included a main effect of depression history and the PiB binding-by-time interaction with effects in the same direction as before (
Table 3). Lower AMNART score was also associated with steeper increases in anxiety-concentration score across time (
Table 3). No other fixed terms were significant. Among random terms, only the intercept showed significant variance and was retained in the final model.
To examine the possibility that the relationship of PiB binding to anxiety-concentration scores was specifically attributable to two GDS items related to concentration (“Is your mind as clear as it used to be?” and “Do you have trouble concentrating?”), we calculated an average anxiety-only score by excluding these two items. In a secondary model analyzing anxiety-only scores, effects were virtually the same as before (for the PiB binding-by-time interaction, p=0.03; for depression history, p=0.02; for fixed effects, R2=0.02; with random terms included, R2=0.7, p<0.0001), except that the AMNART score-by-time interaction term was no longer predictive in this model.
In the model for dysphoria scores, history of depression was associated with a higher adjusted mean for dysphoria scores across time compared with no depression history. No other fixed terms were significant except for a positive linear effect of time indicating greater dysphoria scores over time (for depression history, p<0.0001; for time, p=0.003; for fixed effects, R2=0.04; with random terms included, R2=0.72, p<0.0001). Significant random terms were an uncorrelated intercept and linear slope across time.
For the final model for apathy-anhedonia scores, significant predictors included an interaction of age with time (older age raised the trajectory of apathy-anhedonia scores), an interaction of Hollingshead score with time (lower socioeconomic status lowered the trajectory of symptoms across time), and a main effect of AMNART score (higher cognitive reserve was associated with greater apathy-anhedonia scores across time) (
Table 4). Among random terms, only the intercept showed significant variance and was retained in the final model.
Residuals from predictions of the random and fixed terms for all final models reasonably conformed to assumptions of normality and homogeneity of variance. For the dysphoria model, however, residuals from the fixed-term predictions alone were somewhat positively skewed because of the floor effect of zero values in the distribution of dysphoria scores.
Discussion
We examined the relationship of brain amyloid beta burden to longitudinal measures of depression in a community-based sample of cognitively normal older people and found that higher baseline amyloidosis was associated with worsening depressive symptoms over time. Higher brain amyloid beta levels were associated with increasing anxious-depressive symptom ratings rather than with ratings on symptoms related to dysphoria or apathy-anhedonia in this sample, suggesting that this particular dimension of depressive symptoms may be most useful as an early, dynamic marker in preclinical Alzheimer’s disease.
Our results are consistent with recent findings from two other clinical imaging cohort studies. Investigators at the Washington University Alzheimer’s Disease Research Center reported (
9) that cognitively normal participants with a high baseline amyloid beta burden had greater change scores on the GDS-15 over 1 year compared with those with a low amyloid beta burden, adjusting for age, sex, and education. Similarly, the Australian Imaging, Biomarkers, and Lifestyle research group found (
10) that cognitively normal participants with high amyloid beta levels were over four times more likely to develop categorical depression (defined by the standard GDS-15 cutoff) at 54 months compared with those with low amyloid beta levels, in unadjusted analyses. As in the HABS cohort, these samples included some participants with a depression history and/or antidepressant medication use and GDS scores at baseline were generally low (
9) or in a subclinical range (
10). No significant differences in GDS score or other mood disturbance measures were found between high and low amyloid beta groups at baseline in either study.
While significant cross-sectional associations of brain amyloid beta and depression were not found in these cognitively normal cohorts using amyloid beta–specific PET ligands, Yasuno and colleagues reported (
23) an age- and education-adjusted association of PiB-PET-derived amyloid beta measures and low-range GDS-15 scores among cognitively normal older individuals with elevated PiB retention (only values within the highest two tertiles for the sample were analyzed). This sample excluded individuals with a depression history or antidepressant use, reducing the likelihood of reverse causation and providing indirect support for high amyloid beta levels as a factor that precedes subclinical depressive symptoms.
We provide further biomarker evidence for depression-related symptoms as outcomes of Alzheimer’s pathological changes at the preclinical stage and, in this case, independent of previously diagnosed depression. Regardless of depression history, higher amyloid beta burden predicted de novo or rising depression-spectrum symptoms in the near term that may lead to clinical depression over the longer term. Notably, 7% of the HABS sample reported a current diagnosis of depression, and the unadjusted mean PiB value was significantly higher in these participants compared with those with no depression history. This points to amyloid beta–related brain changes as a possible etiological basis for clinical depression in a subset of these participants. Early expansion of amyloid beta, interacting with tau, within medial temporal lobe structures such as the entorhinal cortex, may affect activity in functionally coupled limbic and neocortical regions, resulting in changes in emotional regulation (
24). The emergence of these symptoms in individuals with high amyloid beta levels may also coincide with progressive amyloid beta deposition and local neurodegeneration within subcortical structures and circuits involved in emotional responses, such as anxiety (
25,
26).
Theoretical constructs of depression and other neuropsychiatric symptoms in neurodegenerative disorders, along with instruments for their measurement, continue to evolve (
27–
29). In line with recent consensus criteria, anxiety may be a symptom of emotional dysregulation in preclinical Alzheimer’s disease that could anticipate syndromal depression or other changes in emotion, temperament, and behavior, as encompassed in the newly defined mild behavioral impairment construct (
29).
Neuropsychiatric symptoms may be most useful as clinical or prognostic markers in cognitively normal older individuals with evidence of other biological risk factors or as sentinels of decline (
30). Holmes and colleagues (
31) found higher anxiety subscale scores, assessed by the Hospital Anxiety and Depression Scale, in APOEε4 carriers compared with noncarriers, specifically within the subgroup of cognitively normal older people with high amyloid beta levels. No main effect of amyloid beta group with anxiety was reported. In a subset of the HABS cohort, we previously reported (
32) that greater amyloid beta burden was associated with higher self-reported loneliness, a novel neuropsychiatric symptom measured by the three-item UCLA Loneliness Scale, after adjustment for anxiety scores on the Hospital Anxiety and Depression Scale and other demographic and psychosocial factors. This effect was also found to be stronger in APOEε4 carriers compared with noncarriers.
We found no main effect of APOEε4 in models for GDS total score or anxiety-concentration scores, and in a post hoc model, no interaction effect of APOEε4 status and amyloid beta level on longitudinal GDS scores. Similarly, Locke and colleagues (
33) found no direct effect of APOEε4 carrier status on longitudinal depression scores in more than 600 cognitively normal adults, ages 21–86, followed for nearly 8 years. Given the low endorsement of depressive symptoms in our cohort and the confounded relationship of amyloid beta level and APOEε4 status, this post hoc analysis may have lacked sufficient power to detect a true relationship between the amyloid beta-by-APOEε4 interaction and the low-range GDS scores in these analyses.
Low educational attainment and income are established risk factors for greater depressive symptom burden in community-dwelling older people (
34,
35), whereas in our models cognitive reserve and socioeconomic status did not predict total GDS scores or anxiety-only scores. This lack of effect may be attributable to the relatively high education and socioeconomic status of the sample, which diminished the influence of these factors. In other results, we found associations of higher cognitive reserve and socioeconomic status with greater apathy-anhedonia scores, controlling for age and time, in the opposite direction to expectations (
35,
36). These results most likely reflect a survivor effect specific to this cognitively normal sample.
It is important to acknowledge that the fixed effects in these models, such as the PiB binding-by-time interaction and depression history, account for a small proportion of the variance for depression scores over time and that the magnitude of this effect also appears to be small. At the same time, it is plausible that the strength of these relationships was attenuated by antidepressant medication use. While these findings suggest that amyloid beta may play a role in the pathogenesis of certain forms of late-life depression, this observed relationship may be limited or indirect. Individuals with the highest levels of amyloid beta are also likely to have tau accumulation and neurodegenerative brain changes that may mediate the reported relationship between higher amyloid beta levels and rising depression scores (
37). If not directly biologically determined, these symptoms may also be a psychological reaction to other subtle cognitive or somatic changes and stresses occurring in late-stage preclinical Alzheimer’s disease. Finally, as multiple and heterogeneous factors may have an impact on depression within an individual and across a sample, psychosocial factors and pathogenic processes such as vascular disease and stress-related mechanisms may prove to be stronger predictors of late-life depressive symptoms than amyloid beta level. Additional studies in both clinically depressed and nonclinical samples are needed to determine whether depressive-spectrum symptoms have sufficient specificity and power to meaningfully inform preclinical Alzheimer’s disease assessment in routine screening or in selected groups.
There are limitations to this study. The relationships observed in these analyses were likely affected by study exclusion criteria, including restricted GDS scores at screening. For example, dysphoric symptoms were infrequently endorsed in this sample, limiting our ability to detect associations with amyloid beta. Individuals with major psychiatric disorders other than mild remitted depression or with active medical and neurological conditions were excluded from the HABS cohort, thereby focusing and limiting the external validity of these findings to older persons with relatively good mental and brain health. Sample size limited our ability to analyze potential moderating effects of antidepressant medications, and, among participants with a depression history, relevant characteristics such as recency of depressive episode and age at onset were not analyzed. Finally, the backward elimination method is sometimes criticized for being too liberal given that multiple runs produce multiple p values. For each dependent variable, however, significant effects found for PiB binding and other predictors of interest in the final model were also significant, or marginally so, in the initial full predictor set. Removal of extraneous terms mostly served to produce parsimonious models.