There is an abundance of evidence that the metabolic glutamate receptor 3 (mGluR3) plays a role in cognition (
1–
7), as well as in the pathophysiology of schizophrenia (
8,
9). Selectively targeting the mGluR3 pathway through its endogenous agonist,
N-acetyl-aspartyl-glutamate (NAAG) (
10), may be a novel strategy for improving cognition in illnesses in which deficits in cognition are prominent (e.g., schizophrenia). NAAG acts as a peptide neurotransmitter that inhibits glutamate neurotransmission as a selective agonist of mGluR3 (
10) and antagonist of N-methyl-
d-aspartate (NMDA) receptors (
11). Agonists of mGluR2/3 have improved cognitive performance and other behaviors in animal models (
12,
13). However, in a phase 2 trial, an mGluR2/3 agonist was not efficacious in treating overall symptoms of schizophrenia (
14). Failure of an mGluR2/3 agonist may be due, in part, to its lack of specificity for mGluR3 (over mGluR2) or differences in downstream signaling from this allosteric modulator or because the effects of targeting mGluR3 may be relatively specific to cognition.
Discussion
There has been great interest in targeting the mGluR3 pathway for the treatment of cognitive dysfunction, and potentially other symptoms of schizophrenia. Targeting the GCPII/FOLH1 enzyme gives an alternative and perhaps more specific approach to modifying glutamate neurotransmission, specifically by modulating levels of its endogenous mGluR3 ligand, NAAG. We exploited genetic variation in FOLH1/GCPII to examine the relationship between FOLH1 expression, NAAG levels, cognition, and neural activity during working memory. One advantage to using genetic variation rather than comparing diagnostic groups is that it enabled us to avoid the inherent confounders between healthy subjects and those with severe mental illness. There are many reasons for an increase or decrease, or no difference, in these measurements between healthy and mentally ill cohorts, including but not limited to antipsychotic medication, smoking, drug abuse, and poor nutrition. Furthermore, as we found with this study, the effects of this missense mutation spans diagnostic groups (e.g., both unaffected/healthy subjects and patients with schizophrenia and psychosis), across multiple levels of biology, suggesting that altering mGluR3 by changing NAAG (via FOLH1/GCPII) may improve cognition in other disorders.
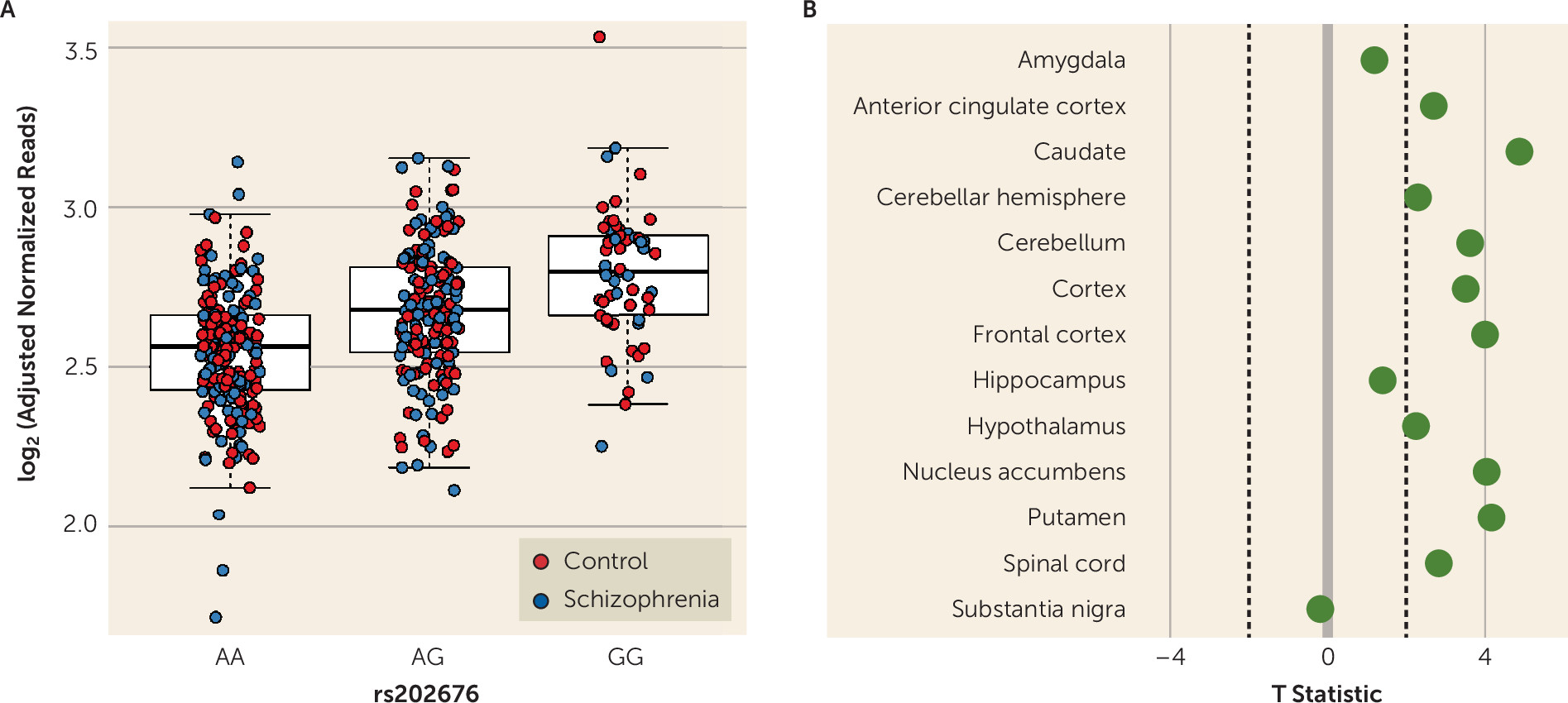
Our data show that among patients with schizophrenia and unaffected control subjects, carriers of the G allele of the FOLH1 missense polymorphism (rs202676) have higher mRNA expression. It should be noted that other genetic variants in linkage disequilibrium with this missense variant also showed strong associations with gene expression, and untangling the specific functional SNPs underlying these associations would be important follow-up molecular work. While we found higher levels of FOLH1 mRNA in carriers of this variant, it was not a direct measure of enzyme function, and therefore we cannot conclusively ascertain the effects of this variant on FOLH1/GCPII enzyme function, which should be explored in future studies.
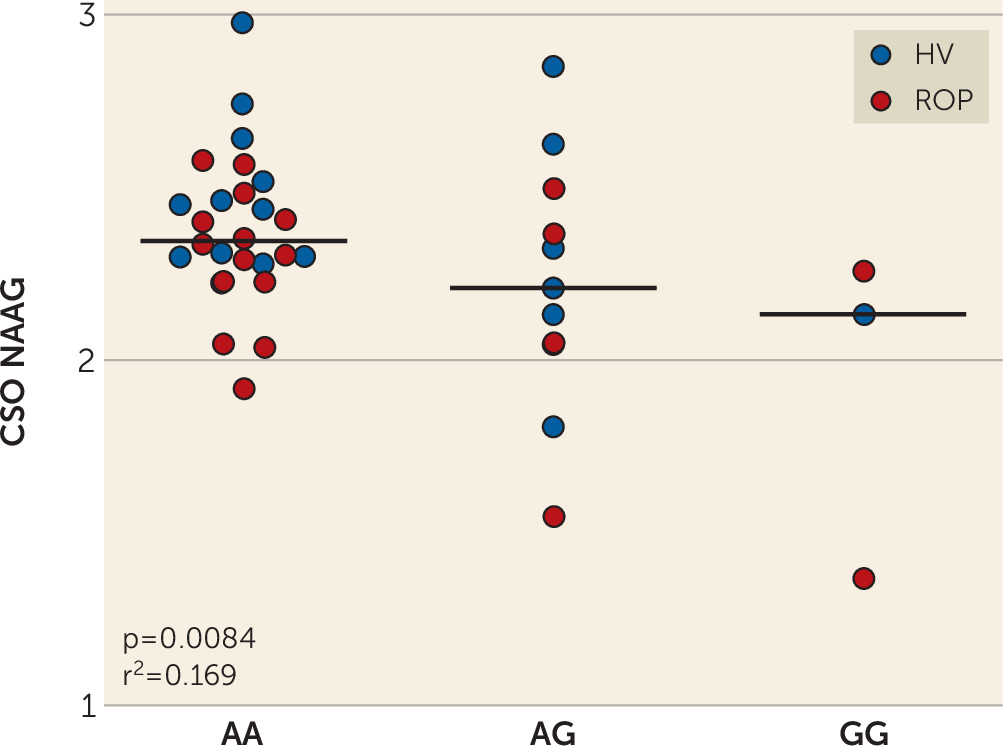
The same mutation associated with higher FOLH1 expression was associated with lower NAAG levels in healthy subjects and in patients with psychosis. Collectively, these findings suggest that decreasing FOLH1/GCPII could increase NAAG levels. These data are the first, to our knowledge, to support such a relationship in human brain. Previous studies comparing NAAG levels in healthy subjects and schizophrenia patients used limited samples, with lower field-strength MRS, and results have been inconsistent. While some studies have found decreased NAAG levels in patients with schizophrenia (
22,
40), other studies have found higher levels (
21,
41) or no differences in NAAG levels (
42), but these studies differed by the brain regions examined and age of the cohort (
22). Additionally, previous research has found both increases and decreases in GCPII expression and NAAG levels between brains of unaffected control subjects and patients with schizophrenia, bipolar disorder, and major depression (
43,
44). In a previous study, our group found that patients with psychosis had decreased NAAG levels compared with healthy control subjects, as measured by 7-T MRS (
27). NAA and NAAG have very similar spectra and thus are hard to separate in in vivo MRS at the widely available field strengths of 1.5 T or 3 T. The present study made use of the increased spectral resolution available at the high magnet field strength of 7 T to provide more accurate estimates of NAAG, separate from the larger signal of NAA. It has been shown previously that NAAG expression is two to four times higher in white matter than in gray matter (
45), and thus the strongest correlation in CSO may, in part, reflect the least uncertainty of the MRS determination in this white matter rich region. Differences in the effect of FOLH1 rs202676 genotype on NAAG levels between the Caucasian and African American cohorts could be due, in part, to differences in allele frequencies and the genetic background on which they are inherited, because CSO NAAG levels are similar between racial cohorts. In the 1000 Genomes Project, the frequency of the G allele was 22.6% in the Caucasian population and 60.2% in the African population (
https://www.ncbi.nlm.nih.gov/projects/SNP). However, in the RNA-sequencing data, FOLH1 mRNA expression was similar in both the Caucasian and African American cohorts, suggesting that the potential feedback of increased expression associated with the missense variant is present in both racial groups and likely relates more to the individual sequence variant than the ancestral background in which it lies. The differences in the clinical data may reflect the influence of other variants on FOLH1 expression or on expression of the pseudogene (FOLH1B) that differs between racial groups.
Genetic variation in FOLH1 was not associated with levels of glutamate or NAA. Levels of glutamate are largely regulated by glutamate transporters. Levels of NAA are regulated by N-acetylaspartate synthase (i.e., NAA synthase) and aspartoacylase (which hydrolyses NAA into aspartate and acetate) (
46), as well as FOLH1/GCPII, and therefore changes in NAA levels may not be predictive of changes in FOLH1/GCPII. NAAG levels are more than threefold lower than NAA levels and are regulated primarily by FOLH1/GCPII and, to a much lesser extent, because of its low abundance, NAAG synthetase (
47); therefore, it is not surprising that genetic variation in FOLH1 influences NAAG levels. In addition, because brain levels of NAA are substantially higher than NAAG levels, modest changes in GCPII hydrolysis of NAAG would have a very small effect on basal NAA levels at best. Future studies should investigate the effects of genetic variation on FOLH1 expression and enzyme activity in both gray and white matter regions.
Animal studies have shown that blocking GCPII results in increases in brain NAAG levels (
17–
19,
48,
49), which is associated with improved cognition (
18,
19) and decreases in schizophrenia-like behaviors (
20,
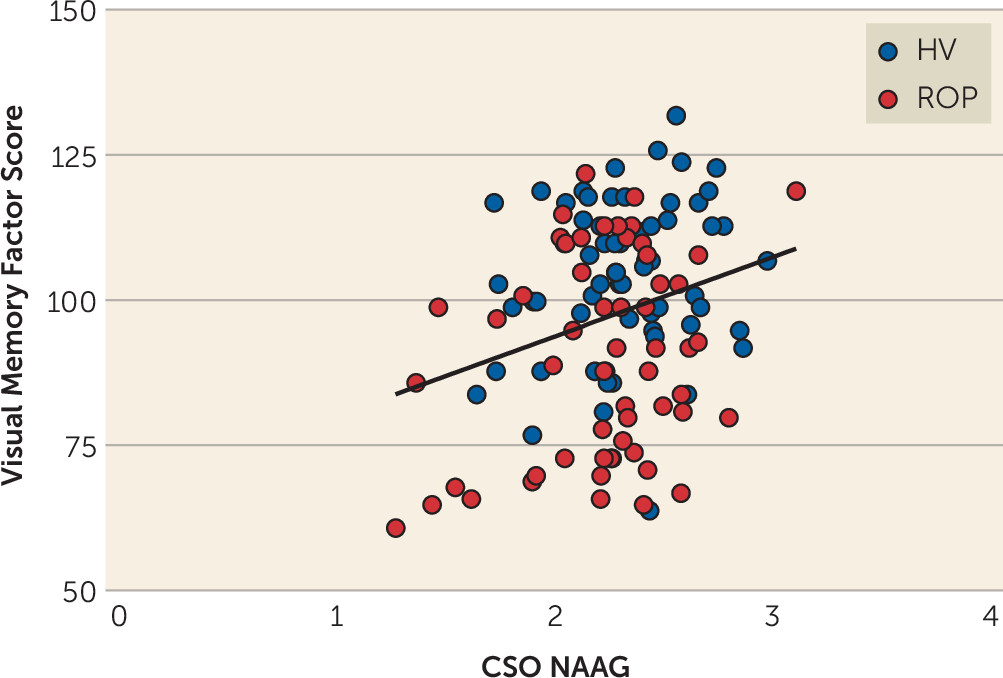
50). However, the effects of differential NAAG levels on cognition have not previously been well studied in patients with schizophrenia or other disorders characterized by cognitive impairment. We found that subjects who had higher levels of NAAG in the CSO performed better on measures of visual memory. The effects of NAAG may be selective to visual memory performance or may correlate with other cognitive domains that we did not measure or measured in a way that did not capture the effect. It is interesting to note that the
N-back task is a visual (working) memory task, similar to the tests used in the visual memory domain in the clinical cohort. Future studies will need to explore the effects of CSO NAAG levels on other cognitive tests, including other cognitive tasks during fMRI, in order to more conclusively determine the level of specificity, or not, of our findings in the visual memory domain. NAAG levels in CSO may correlate with visual memory because this measurement reflects general NAAG levels in cortical white matter and thus widespread integrity of the connections needed throughout the cortex for visual memory, whereas measurement of NAAG in other regions may correlate with other cognitive domains. Similarly, other investigators have reported a positive correlation between frontal lobe NAAG and NAAG/NAA ratio and measures of episodic memory in patients with schizophrenia (
51), although they also reported higher NAAG levels in the ACC in patients with schizophrenia (N=20) compared with healthy subjects (N=20) (
51). One study found that multiple sclerosis patients with cognitive impairment had lower hippocampal NAAG levels than multiple sclerosis patients without cognitive impairment (
18) and a positive correlation between hippocampal NAAG/creatine and cognitive performance in multiple domains (
18).
Here, we focused on the region providing the best NAAG measures (i.e., the CSO), because we did not have a specific hypothesis about a particular region, given that we were testing the effects on IQ and a composite score for cognition, as well as individual domains. Future studies will need to determine the effects of regional NAAG levels on cognition and symptoms of schizophrenia. NAAG levels in the DLPFC were positively associated with visual memory scores, although this finding may be less significant than associations with CSO NAAG, in part as a result of the greater variance in the NAAG estimates in the DLPFC. Recent data on monkeys have shown that mGluR3 receptors have an expansive role in primate DLPFC circuits governing cognition (
4). In primates, mGluR3 receptors are expressed on both astrocytes and postsynaptically in spine synapses in the DLPFC (
4). Increasing NAAG levels strengthens connectivity and greatly enhances persisting firing of DLPFC Delay cells needed for higher cognition (
4). These data may be the mechanism behind our data showing that higher NAAG levels result in better cognitive performance in humans.
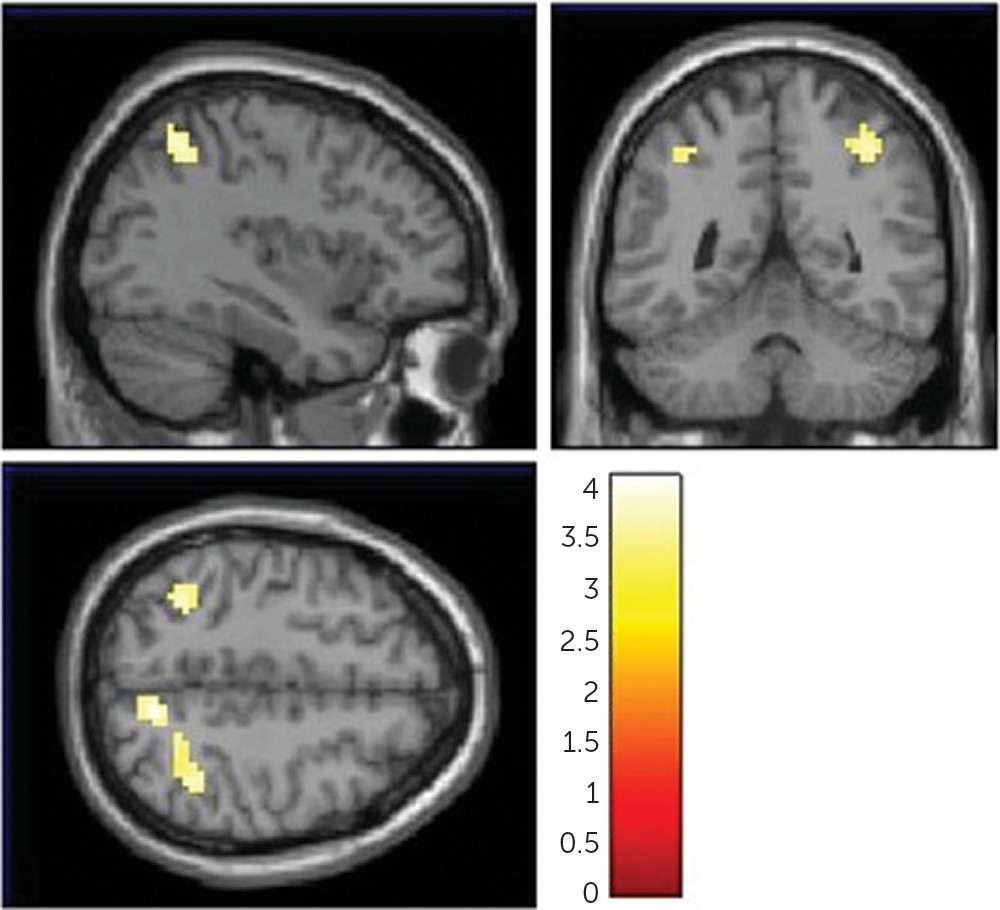
In order to determine the neural basis underlying the association between NAAG and working memory, we correlated FOLH1 genetics with cortical efficiency during working memory, using fMRI. The
N-back working memory task (
52) has been widely applied to study working memory dysfunction in schizophrenia (
22,
23) and strongly engages a working memory neural network that includes the DLPFC and parietal cortex (
53,
54) in a capacity-constrained response function (inverted U) (
52). We found that carriers of this FOLH1 missense mutation had higher cortical activity during working memory. Because performance was similar between subjects (>80% accuracy) and did not differ between genotype groups, the working memory activity associated with the G allele was interpreted as inefficient (suboptimal) (
23), because greater resources (activity) were required to reach the same level of performance. Increased cortical activity during working memory is a pattern of brain activation found in patients with schizophrenia (
22) and their healthy siblings (
23). These genetic-related inefficient activity patterns overlap with inefficient working memory responses from several other studies that found parietal, cuneus, and superior temporal hyperactivation during working memory in schizophrenia patients compared with healthy subjects (
55–
57). We have also shown that healthy subjects who carry SNPs associated with risk for schizophrenia have greater cortical activity during working memory, for genes including GRM3 (
3), COMT (
37), and CACNA1C (
58). When investigating the correlation between genotype and working memory fMRI signals, it is common to find significance in a subset of the regions (
58–
60), rather than the entire network, as is the case here. While the block-design fMRI
N-back task is advantageous for robust signals, it is difficult to make concrete functionality conclusions beyond those related to general working memory processes. Additionally, one study found that across a series of memory tasks, there was a linear increase in brain activity (measured by fMRI) with increasing age in areas that were normally decreased during task performance (e.g., the medial frontal and parietal regions), whereas activity in regions with task-related activation (e.g., the dorsolateral prefrontal cortex) decreased with age (
61), suggesting a gradual, age-related reduction in the ability to suspend non-task-related or default-mode activity and engage areas involved in carrying out memory tasks. Cognitive dysfunction related to increased age may be similar in this way to what we have shown in patients with schizophrenia (
10,
33,
34), shown by a shift in the balance between DLPFC activity and parietal activity, which could account for increased vulnerability to distraction from irrelevant information (
61). This pattern of brain activity is consistent with weaker DLPFC connectivity (
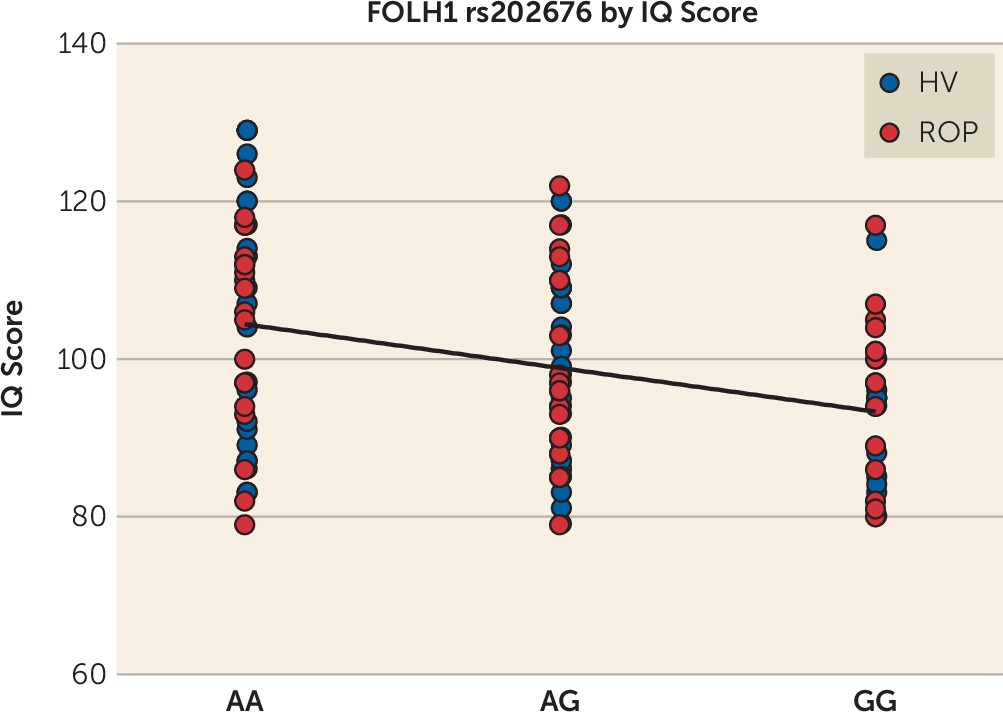
4) and a need for a more posterior cortical strategy (e.g., hyperactivity of the parietal cortex) to maintain a high working memory performance. In the present study, we found that healthy and psychotic subjects who are carriers of the FOLH1 variant that is associated with lower NAAG levels had a lower IQ score, which suggests that low NAAG levels could result in widespread cognitive dysfunction.
In addition to the cognitive effects reported here, this missense mutation has been associated with less severe negative symptoms (
62) and greater reduction in negative symptoms following folic acid and vitamin B
12 supplementation (
63). Therefore, genetic variation in FOLH1 and related expression and NAAG concentration differences may be relevant not only to cognition in schizophrenia (and related potential therapeutics) but also to negative symptoms in schizophrenia (
64). Future studies should test the relationship between NAAG levels and genetic variation in FOLH1 on negative symptoms in schizophrenia.
Consistent with the implications of these findings, previous research has suggested that increasing NAAG levels can improve cognition. One study showed that treatment with growth hormone-releasing hormone (GHRH) increases brain levels of NAAG and GABA, but not glutamate levels, in the DLPFC in older adults with mild cognitive impairment and cognitively intact older adults (
65). The study also found an improvement in the cognitive domains of executive function and verbal memory, which may be related to the increased NAAG and/or GABA levels (
65) or another aspect of GHRH actions. Inhibitors of GCPII/FOLH1 have been shown to increase neural NAAG levels and improve learning and memory in animal models (
18,
19) but are not currently available for clinical use. We focused on NAAG because it is a substrate for mGluR3 (unlike NAA), and NAAG levels are primarily regulated by FOLH1/GCPII. We found that CSO NAA levels are not associated with cognitive performance, but the ratio of NAAG to NAA is associated with visual memory performance.
Before introducing a GCPII antagonist into a patient population, it is imperative to fully ascertain all the nuances of all the variables (e.g., cognitive domain, metabolite measurements, sample size), considering not only our findings but also synthesizing relevant findings from previous as well as future studies. It is important to develop a better understanding of how modulating NAAG levels (e.g., with a GCPII inhibitor) (
20,
50,
66–
68) will affect cognitive performance and functional neural activity in humans. Our study provides insight regarding the neurobiological impact of NAAG modulation, including which circuits are most likely to be affected and could serve as biomarkers in clinical trials of GCPII inhibitors. Measuring NAAG levels using MRS, cognitive function, and neural activation using fMRI in a battery of tasks in a large cohort of the same patients will be prudent. Additionally, future studies should test the relationship between differential FOLH1/GCPII and NAAG results and measures of cognition and/or other symptoms and behaviors in patients with schizophrenia, as well as other causes of cognitive impairment and/or decline.