Bipolar I and bipolar II disorders are serious mental illnesses associated with a wide array of debilitating symptoms, including episodes of mania, hypomania, and depression (
1). Depressive episodes in bipolar I and II disorders (bipolar depression) are more prevalent than episodes of mania or hypomania in most patients and are associated with greater disability and decreased quality of life (
1). Currently, the second-generation antipsychotics cariprazine, quetiapine (and extended-release quetiapine), lurasidone, and olanzapine in combination with fluoxetine are approved for the treatment of depressive episodes in bipolar I disorder (
2–
6). Treatment options for depression associated with bipolar II disorder are even more limited, with only quetiapine (and extended-release quetiapine) approved for treatment (
5,
7).
Approved antipsychotics for bipolar depression are associated with a range of undesirable side effects, including cardiometabolic disturbances, motor impairments, and hyperprolactinemia (
8). These adverse effects are a major contributor to nonadherence with antipsychotic treatment (
9,
10). In addition, the use of psychotropic medications for bipolar disorder, including antidepressants and antipsychotics, is associated with increased risk for type 2 diabetes mellitus (
11), metabolic syndrome (
12), cardiovascular disease (
13), obesity, and movement and seizure disorders (
14), which exacerbate the already increased risk of cardiovascular disease, coronary heart disease, and cerebrovascular disease associated with severe mental illness (
13). The use of antidepressants in depressed patients with bipolar disorder is of uncertain value and is associated with potential switching into mania in bipolar I disorder (
2). Thus, a new treatment option that is effective for depressive episodes in both bipolar I and bipolar II disorders and has a more benign and favorable safety profile could improve patient outcomes, with lower morbidity and a higher quality of life.
Lumateperone (lumateperone tosylate), a mechanistically novel antipsychotic, is approved by the U.S. Food and Drug Administration (FDA) for the treatment of schizophrenia (
15). Lumateperone simultaneously modulates serotonin, dopamine, and glutamate neurotransmission, the key neurotransmitters implicated in serious mental illnesses (
16–
18). Lumateperone functions as a potent serotonin 5-HT
2A receptor antagonist, a dopamine D
2 receptor presynaptic partial agonist and postsynaptic antagonist, a D
1 receptor–dependent modulator of glutamate, and a serotonin reuptake inhibitor (
16,
17). These properties, combined with lack of interaction with receptors that contribute to cardiometabolic side effects associated with other antipsychotic medications (
17), make lumateperone an attractive candidate for the treatment of mood disorders. In late-phase controlled clinical trials in schizophrenia, lumateperone treatment for up to 4 weeks was effective without significant extrapyramidal, cardiometabolic, or endocrine side effects compared with placebo (
19,
20). The favorable safety profile of 42 mg/day of lumateperone in schizophrenia was confirmed for up to 1 year of treatment in an open-label clinical trial (
21).
In this multinational randomized double-blind placebo-controlled phase 3 study, we evaluated the efficacy and safety of lumateperone for the treatment of major depressive episodes associated with bipolar I and bipolar II disorders.
METHODS
Patients
Eligible participants were 18 to 75 years old, with a confirmed diagnosis of bipolar I or bipolar II disorder according to DSM-5, who were experiencing a major depressive episode. Patients were required to have depression of at least moderate severity, with a total score ≥20 on the Montgomery-Åsberg Depression Rating Scale (MADRS) (
22) and scores ≥4 on the depression and overall bipolar illness subscales of the Clinical Global Impressions Scale–Bipolar Version severity scale (CGI-BP-S) (
23) at screening and baseline. The duration of the major depressive episode must have been at least 2 weeks but less than 6 months before screening, and symptoms must have caused clinically significant distress or functional impairment. Patients were required to have a score ≤12 on the Young Mania Rating Scale (YMRS) (
24) at screening and baseline. Patients were recruited from the clinical practices of participating investigators or via institutional review board–approved recruitment materials to identify potential participants in their catchment areas.
Patients were excluded if they had a decrease ≥25% in MADRS score between screening and baseline, had a significant risk for suicidal behavior, or had been diagnosed with a psychiatric illness other than bipolar disorder within 12 months of screening. Additional inclusion and exclusion criteria are listed in the
online supplement.
All patients provided written informed consent as approved by the responsible institutional review board or independent ethics committee before participating in any study-related activities.
Study Design, Intervention, and Randomization
This was a 6-week multicenter randomized double-blind placebo-controlled outpatient study (NCT03249376) conducted at 54 clinical sites in six countries: the United States (14 sites), Bulgaria (10 sites), Colombia (three sites), the Russian Federation (11 sites), Serbia (five sites), and Ukraine (11 sites). During a screening period of up to 2 weeks, patients eligible for participation discontinued their current antidepressant or other psychotropic treatment. At baseline, patients stratified by bipolar I or bipolar II diagnosis were randomized in a 1:1 ratio to receive treatment with either 42 mg/day of lumateperone (equivalent to 60 mg/day of lumateperone tosylate) or placebo. Patients were randomized using an interactive voice or web response system. Independent biostatistics personnel not participating in the conduct of the study generated a permuted block randomization schedule for the interactive system, linking sequential patient randomization numbers to treatment codes. Lumateperone was administered via capsule, with or without food, once daily in the evening for 6 weeks. Safety and efficacy assessments were conducted at weekly clinic visits (days 8, 15, 22, 29, 36, and 43) and at a final safety follow-up visit approximately 2 weeks after the last dose of study medication. Study medication adherence was calculated as the percentage of adherent days during the treatment period. Adherent days were defined as days during the treatment period on which a patient took one capsule of study medication.
This study was performed in accordance with the principles outlined in the Declaration of Helsinki and in compliance with Good Clinical Practice guidelines.
Measures and Procedures
The primary and key secondary endpoints were the efficacy of 42 mg/day of lumateperone compared with placebo, measured by mean change from baseline to day 43 in MADRS total score and CGI-BP-S total score, respectively. CGI-BP-S total score was calculated as the sum of the CGI-BP-S subscores for depression, mania, and overall bipolar illness; the individual CGI-BP-S subscores were also evaluated. Additional efficacy measures included response to treatment (defined as a decrease ≥50% in MADRS score), remission (defined as a MADRS score ≤12), improvement in MADRS and CGI-BP-S scores by week of treatment, and percent score on the Quality of Life Enjoyment and Satisfaction Questionnaire–Short Form (Q-LES-Q-SF) (
25). Safety was assessed by incidence of treatment-emergent adverse events (coded according to the Medical Dictionary for Regulatory Activities, version 20.1), clinical laboratory evaluations, ECG, physical and neurological examination, and vital sign measurements. Extrapyramidal symptoms were assessed by the Simpson-Angus Scale (
26), the Barnes Akathisia Rating Scale (
27), and the Abnormal Involuntary Movement Scale (
28). Mania was monitored using the YMRS, and suicidality was evaluated with the Columbia–Suicide Severity Rating Scale (C-SSRS) (
29).
Statistical Analysis
Treatment effects on primary and key secondary efficacy endpoints were evaluated using a mixed-effects model for repeated measures in the prespecified modified intent-to-treat population, defined as all patients who received at least one dose of study medication and had a valid baseline MADRS assessment and at least one valid postbaseline MADRS assessment. The model included visit, treatment group, site, and bipolar disorder stratification (bipolar I or bipolar II disorder) as factors. The patient term was included as a random effect, and baseline score was included as a covariate, with interaction terms for treatment group-by-visit and visit-by-baseline score. An unstructured covariance matrix was used to estimate the correlation of repeated measurements within a patient. Sensitivity analyses for primary and key secondary endpoints used an analysis of covariance (ANCOVA), with missing data imputed using the last observation carried forward. To control the type I error rate for multiple comparisons of the primary and key secondary efficacy parameters, a fixed-sequence hierarchical gatekeeping strategy with a two-sided significance level of 0.05 was used.
Safety parameters were summarized descriptively by treatment group and visit in the safety population, defined as patients receiving at least one dose of study drug. Laboratory assessment summaries included by-visit and change from baseline values and incidence of patients meeting markedly abnormal criteria. An exploratory analysis compared differences in prespecified clinical chemistry parameters between the lumateperone and placebo groups.
In each treatment arm, 163 patients were expected to have evaluable data. The study was designed to have 85% power to demonstrate a clinically relevant treatment difference from placebo of 3 points in MADRS score, with a common standard deviation of 9.0, at a two-sided significance level of 0.05. Statistical analyses were performed with SAS, version 9.4 or higher (SAS Institute, Cary, N.C.).
RESULTS
Patient Population
Of the 546 patients screened for eligibility, 381 were randomized (lumateperone, N=191; placebo, N=190), and 377 received treatment and were included in the safety population (see Figure S1 in the
online supplement). The average time from screening to randomization was 14.5 days (SD=5.18). There were 376 patients in the modified intent-to-treat efficacy population (lumateperone, N=188; placebo, N=188); 333 patients completed the 6-week treatment period (lumateperone, N=167; placebo, N=166). The most common causes of discontinuation from treatment were adverse events (lumateperone, 5.8%; placebo, 2.6%) and patient withdrawal of consent (1.6% and 4.7%, respectively) (see
Figure S1).
Baseline demographic and clinical characteristics were similar between the lumateperone and placebo treatment groups (
Table 1). The majority of patients were White (91.2%) and had bipolar I disorder (79.8%). The overall population had moderate to severe depression symptoms at baseline, as indicated by a mean baseline MADRS score of 30.5 and a mean CGI-BP-S depression subscore of 4.6 (
30). The mean age at first bipolar diagnosis was 32.6 years (range, 5–63 years). Prior to treatment with study drug, 49.9% of patients were being treated with antipsychotics, antidepressants, and/or mood stabilizers; 50.1% of patients were not treated with these prior medications. In the modified intent-to-treat population, treatment adherence was 99.7% for both groups, and none of the patients were nonadherent, defined as <80% or >120% adherence.
Efficacy
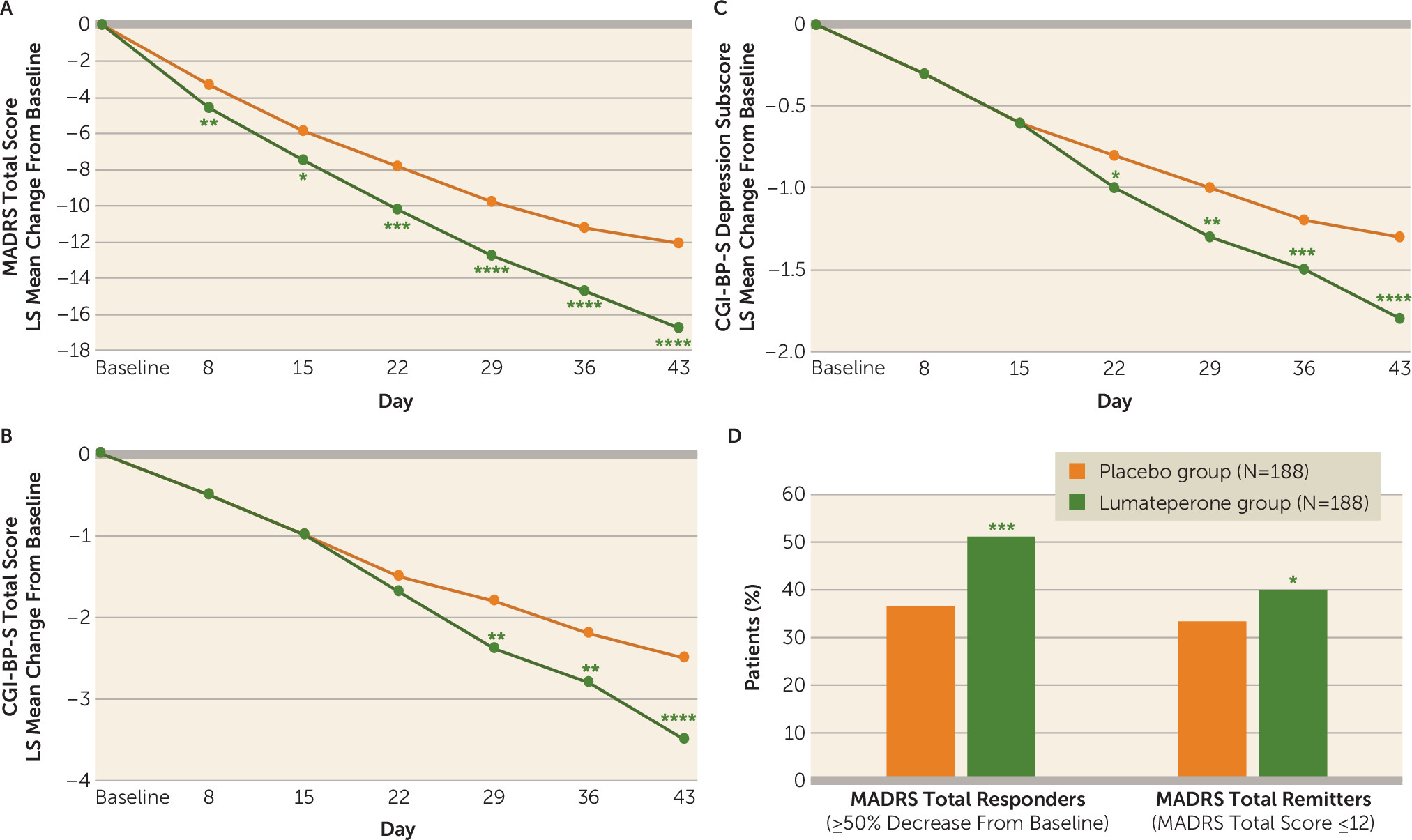
Lumateperone treatment was associated with a statistically significant greater reduction in MADRS score from baseline to day 43 compared with placebo (least squares [LS] mean change, −16.7; LS mean difference compared with placebo, −4.6, 95% CI=−6.34, −2.83; effect size=−0.56, p<0.0001) (
Figure 1A). Lumateperone significantly improved MADRS score compared with placebo as early as day 8, with continuing improvement throughout the study (
Figure 1A). Improvement in MADRS score at day 43 with lumateperone was supported by an ANCOVA last-observation-carried-forward sensitivity analysis (LS mean difference, −4.7, 95% CI=−6.4, −3.0; effect size=−0.55, p<0.001,
Table 2). Treatment with lumateperone resulted in significantly greater rates of response at day 43 compared with placebo (51.1% and 36.7%, respectively; p<0.001). Remission rates were also significantly higher at day 43 in the lumateperone group compared with the placebo group (39.9% and 33.5%, respectively; p=0.018).
There was significant improvement in the key secondary efficacy endpoint, change from baseline to day 43 in CGI-BP-S total score for the lumateperone group compared with the placebo group (LS mean change, −3.5; LS mean difference, −0.9, 95% CI=−1.37, −0.51; effect size=−0.46, p<0.0001) (
Figure 1B). ANCOVA last-observation-carried-forward sensitivity analysis of CGI-BP-S total score supported the robustness of the primary analysis (LS mean difference, −1.0, 95% CI=−1.46, −0.61; effect size=−0.49, p<0.001) (
Table 2). At day 43, lumateperone treatment compared with placebo was also associated with significantly improved CGI-BP-S subscores for depression (LS mean difference, −0.5, 95% CI=−0.75, −0.30; effect size=−0.50, p<0.0001) (
Figure 1C) and for overall bipolar illness (LS mean difference, −0.4, 95% CI=−0.65, −0.22; effect size=−0.43, p<0.0001) (
Table 2). Change from baseline to day 43 in CGI-BP-S mania subscore was minimal and similar to placebo (LS mean change, 0.0; LS mean difference, −0.0, 95% CI=−0.08, 0.04; effect size=−0.08, p=0.4). The Q-LES-Q-SF percent score was also significantly improved at day 43 in the lumateperone group compared with the placebo group (ANCOVA LS mean difference, 4.6, 95% CI=1.42, 7.69; effect size=0.31, p=0.005).
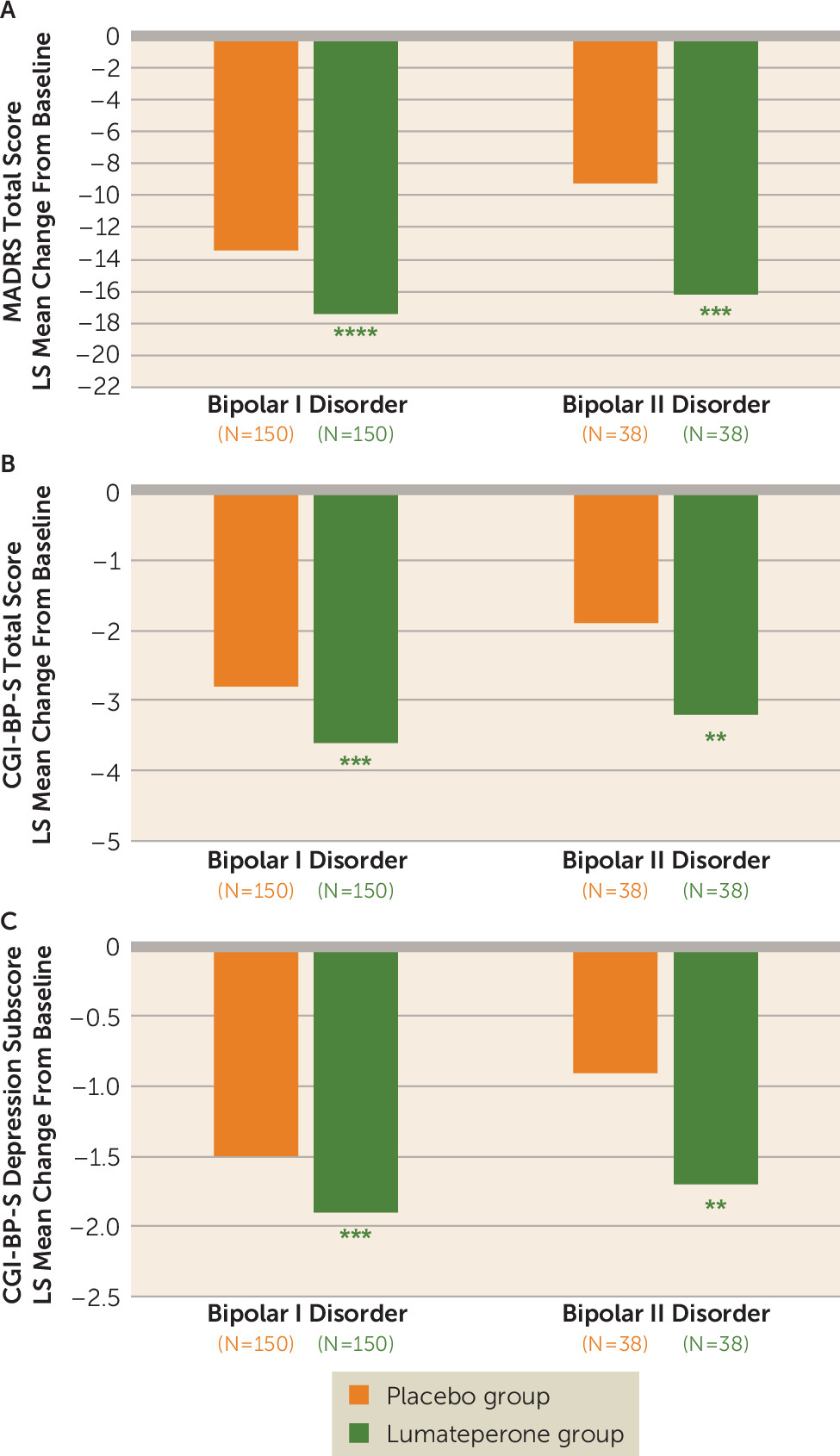
Significant improvement in MADRS score in the lumateperone group compared with the placebo group at day 43 was observed both in patients with bipolar I disorder (LS mean difference, −4.0, 95% CI=−5.92, −1.99; effect size=−0.49, p<0.0001) and in those with bipolar II disorder (LS mean difference, −7.0, 95% CI=−10.92, −3.16; effect size=−0.81, p<0.001) (
Figure 2A). There was also significant improvement in CGI-BP-S total score compared with placebo at day 43 in patients with bipolar I disorder (LS mean difference, −0.9, 95% CI=−1.34, −0.37; p<0.001) and bipolar II disorder (LS mean difference, −1.3, 95% CI=−2.25, −0.34; p<0.01) (
Figure 2B). Significant improvement in CGI-BP-S depression subscore compared with placebo at day 43 was also observed in patients with bipolar I disorder (LS mean difference, −0.5, 95% CI=−0.72, −0.21; p<0.001) and bipolar II disorder (LS mean difference, −0.8, 95% CI=−1.25, −0.26; p<0.01) (
Figure 2C). Consistent treatment effects were observed for MADRS score and CGI-BP-S total score in subgroups of sex, age (≤40 years and >40 years), and age at illness onset (<22 years and ≥22 years). Significant improvements in MADRS score in patients in the lumateperone group compared with the placebo group were observed at clinical sites located both in the United States (LS mean difference, −3.4, 95% CI=−6.83, −0.02; p<0.05) and outside the United States (LS mean difference, −5.2, 95% CI=−7.25, −3.09; p<0.001).
Safety
The rate of treatment-emergent adverse events occurring with lumateperone (54.8%) was similar to the rate with placebo (50.3%). Drug-related treatment-emergent adverse events occurred in 41.5% of the lumateperone group and 31.2% of the placebo group. The only treatment-emergent adverse events occurring in the lumateperone group in at least 5% of patients and at more than twice the rate of the placebo group were somnolence (lumateperone, 8.5%; placebo, 1.1%) and nausea (lumateperone, 6.4%; placebo, 2.1%). The majority of treatment-emergent adverse events were mild to moderate in severity, with four patients (2.1%) in the lumateperone group experiencing severe treatment-emergent adverse events, including insomnia (two patients, 1.1%), headache (one patient, 0.5%), and somnolence (one patient, 0.5%). Treatment-emergent adverse events led to discontinuation of 11 patients (5.9%) in the lumateperone group and four patients (2.1%) in the placebo group. Treatment-emergent adverse events that led to discontinuation of at least one patient were mania (two patients [1.1%] in each group) and insomnia (two patients [1.1%] in the lumateperone group). The proportion of patients experiencing mania was low in both groups (lumateperone, 1.1%; placebo, 2.1%). Additionally, there was one case of hypomania (0.5%) in each group. There was one treatment-emergent serious adverse event of mania in the lumateperone group, which led to discontinuation. There was no worsening of mania in either group as measured by mean change from baseline to day 43 in YMRS score (lumateperone, −1.4; placebo, −0.9). Nine patients (2.4%) had a YMRS score ≥15 at any point during the study, with a similar proportion between groups (lumateperone, four patients [2.1%]; placebo, five patients [2.7%]).
There was no suicidal behavior in either group during treatment, as assessed with the C-SSRS. Baseline C-SSRS suicidal ideation was reported in 4.3% of patients in the lumateperone group and 7.9% of patients in the placebo group. C-SSRS–assessed suicidal ideation at any time during treatment was reported in 5.3% of patients in the lumateperone group and 10.1% of patients in the placebo group. No patients died during the study. In the modified intent-to-treat population, as-needed zolpidem treatment for insomnia was reported in 1.6% of the lumateperone group and 3.2% of the placebo group.
The only extrapyramidal symptom–related treatment-emergent adverse event was one case (0.5%) of mild dyskinesia in the lumateperone group, which started on day 43 and was considered drug-related by the investigator. Per protocol, the final dose of lumateperone was on day 42. This patient had a history of tardive dyskinesia. There were no significant changes from baseline in Barnes Akathisia Rating Scale, Abnormal Involuntary Movement Scale, or Simpson-Angus Scale scores in either group. Concomitant benzodiazepine use was permitted and was reported in four patients in the lumateperone group (2.1%) and 10 patients in the placebo group (5.3%) during the study.
Minimal changes in weight and body morphology were observed in both groups (
Table 3). Potentially clinically significant weight decrease (≥7% decrease from baseline) occurred in 1.1% of patients in the lumateperone group and in none of the patients in the placebo group. In both treatment groups, 1.1% of patients had potentially clinically significant weight increase (≥7% increase from baseline). There were no notable changes in cardiometabolic parameters, including in fasting levels of glucose, total cholesterol, high-density lipoprotein cholesterol, low-density lipoprotein cholesterol, triglycerides, and insulin (
Table 3). There were no notable changes in endocrine parameters and no increases in prolactin in either treatment group (
Table 3). No patients had a QTc (Fridericia corrected) interval >500 ms at any time; rates of an increase of ≥60 ms from baseline were low and were similar between the lumateperone (one patient, 0.6%) and placebo (three patients, 1.8%) groups.
DISCUSSION
In this multinational randomized double-blind placebo-controlled study, treatment with lumateperone monotherapy at 42 mg/day was significantly associated with improved symptoms in major depressive episodes in patients with bipolar I or bipolar II disorder. Treatment with lumateperone was associated with a rapid and significant improvement in MADRS score by week 1 (at the first postdose assessment), with continuing improvements throughout the 6-week study. The efficacy of lumateperone on MADRS score, the primary endpoint, was supported by improvements in CGI-BP-S total score, the key secondary endpoint. Sensitivity analyses based on ANCOVA with last observation carried forward confirmed the robustness of the mixed-effects model for repeated measures approach for MADRS score, and no demographic subgroup appeared to drive the overall efficacy. Significant improvement in MADRS score was observed in patients treated both at U.S. study sites and at sites in other countries.
The efficacy of lumateperone in improvement of depression symptoms is similar to that of approved antipsychotic therapies for bipolar I and bipolar II depression. The overall reduction and the placebo-adjusted reduction in MADRS score for lumateperone treatment (mean change, −16.7; LS mean difference, −4.6) was similar to that reported in trials of approved monotherapies for bipolar disorder (mean change range, −13.7 to −19.6; LS mean difference range, −2.5 to −4.8) (
31–
34). The MADRS score effect size for lumateperone treatment compared with placebo was favorable at day 43 (−0.56).
Patient-level improvements supported the clinical relevance of lumateperone treatment. MADRS response rates for lumateperone (51.1%) were comparable to those reported for other FDA-approved treatments (MADRS response rate range, 39% to 65%) (
31–
33). Significantly greater remission rates for lumateperone compared with placebo further support the clinical efficacy of lumateperone. The significant improvements measured by MADRS score were also accompanied by clinically meaningful improvements in quality of life as measured by the Q-LES-Q-SF, which includes assessment of family and social relationships as well as overall sense of well-being.
Lumateperone treatment was effective in patients with both bipolar I and bipolar II disorders (MADRS score effect sizes, −0.49 in bipolar I disorder and −0.81 in bipolar II disorder). In patients with bipolar II disorder, the MADRS score effect size with lumateperone treatment compared favorably with that of quetiapine treatment (−0.45 to −0.54) (
34). Improvements in patients with bipolar II disorder were supported by significant improvements in CGI-BP-S total score and CGI-BP-S depression subscore. While this initial study of lumateperone had a relatively small number of participants with bipolar II disorder (38 per treatment group), improvements with lumateperone are notable, as quetiapine (and its extended-release formulation) is the only antipsychotic currently approved as a monotherapy for depressive episodes associated with bipolar II disorder (
34). However, quetiapine is also associated with a high burden of side effects, including extrapyramidal symptoms, moderate weight gain, sedation, and risk of metabolic syndrome (
5,
7,
8,
35).
Treatment with 42 mg/day of lumateperone for 6 weeks in patients with bipolar I or bipolar II disorder with an associated major depressive episode was well tolerated. In this study, treatment-emergent adverse events were predominantly mild to moderate in severity. Somnolence and nausea were the only adverse events in the lumateperone group that occurred at a clinically meaningful rate. There were no differences between the lumateperone and placebo groups in the incidence of either treatment-emergent mania or suicidal ideation, with a single serious adverse event of mania during treatment. No new safety signals were detected in patients with bipolar disorder. This safety profile is consistent with that of lumateperone at 42 mg/day for the treatment of schizophrenia in both short- and long-term clinical trials (
19–
21).
While lack of tolerability, often due to extrapyramidal symptoms and weight gain, is cited as a major driver of antipsychotic nonadherence (
8), lumateperone was not associated with extrapyramidal symptoms. There was only one case of mild dyskinesia, in a patient who had a history of extrapyramidal symptoms and oral dyskinesia, with exacerbation of dyskinesia and exacerbation of extrapyramidal symptoms reported after completion of day 42 of lumateperone treatment. There was no significant increase from baseline with lumateperone treatment in Barnes Akathisia Rating Scale, Abnormal Involuntary Movement Scale, and Simpson-Angus Scale scores at day 43. In comparison to no incidences of akathisia reported for lumateperone in this study, higher rates of akathisia have been reported with quetiapine (1.5%–4%), lurasidone (8%–11%), and cariprazine (6%–10%) (
4–
7) in clinical bipolar depression trials. Weight, waist circumference, and body mass index were also stable for the duration of this short-term study of lumateperone treatment. As patients with bipolar disorder are vulnerable to cardiovascular disease and metabolic syndrome (
12,
36), the cardiometabolic profile of lumateperone is an important consideration when selecting treatment. The lumateperone treatment group in this study had no meaningful increases in levels of triglycerides, cholesterol, insulin, or glucose, suggesting that it is not associated with an increased risk of metabolic syndrome. Additionally, there was no evidence of hyperprolactinemia in the study, which is associated with many second-generation antipsychotics (
8). The safety profile of lumateperone may be due to its unique mechanism of action, with minimal binding to histaminergic or muscarinic receptors (
17), which have been associated with cardiometabolic effects and other tolerability issues of existing antipsychotics (
37).
Limitations of the study include the exclusion of patients with treatment-resistant illness, imminent suicidal risk, rapid cycling, or serious comorbid psychiatric or medical illnesses, which may limit the generalizability of the findings. This study only assessed lumateperone at 42 mg/day, so dose-response characteristics cannot be established, and it did not include an active treatment arm, so comparisons with other therapies are historical. Lastly, the safety data in this study are for short-term exposure; additional studies are needed to examine long-term safety in patients with bipolar disorder. Of note, lumateperone had a favorable safety and tolerability profile in a 1-year study in patients with stable schizophrenia (
21).
In summary, 42 mg/day of lumateperone significantly improved depression in patients with bipolar I or bipolar II disorder experiencing an acute major depressive episode. Six-week treatment with lumateperone was generally well tolerated, with low risk for extrapyramidal symptoms and minimal adverse effects on metabolic parameters, prolactin, or weight. Lumateperone’s clinical profile indicates that it is a promising treatment option for major depressive episodes associated with bipolar I or bipolar II disorder.