Discussion
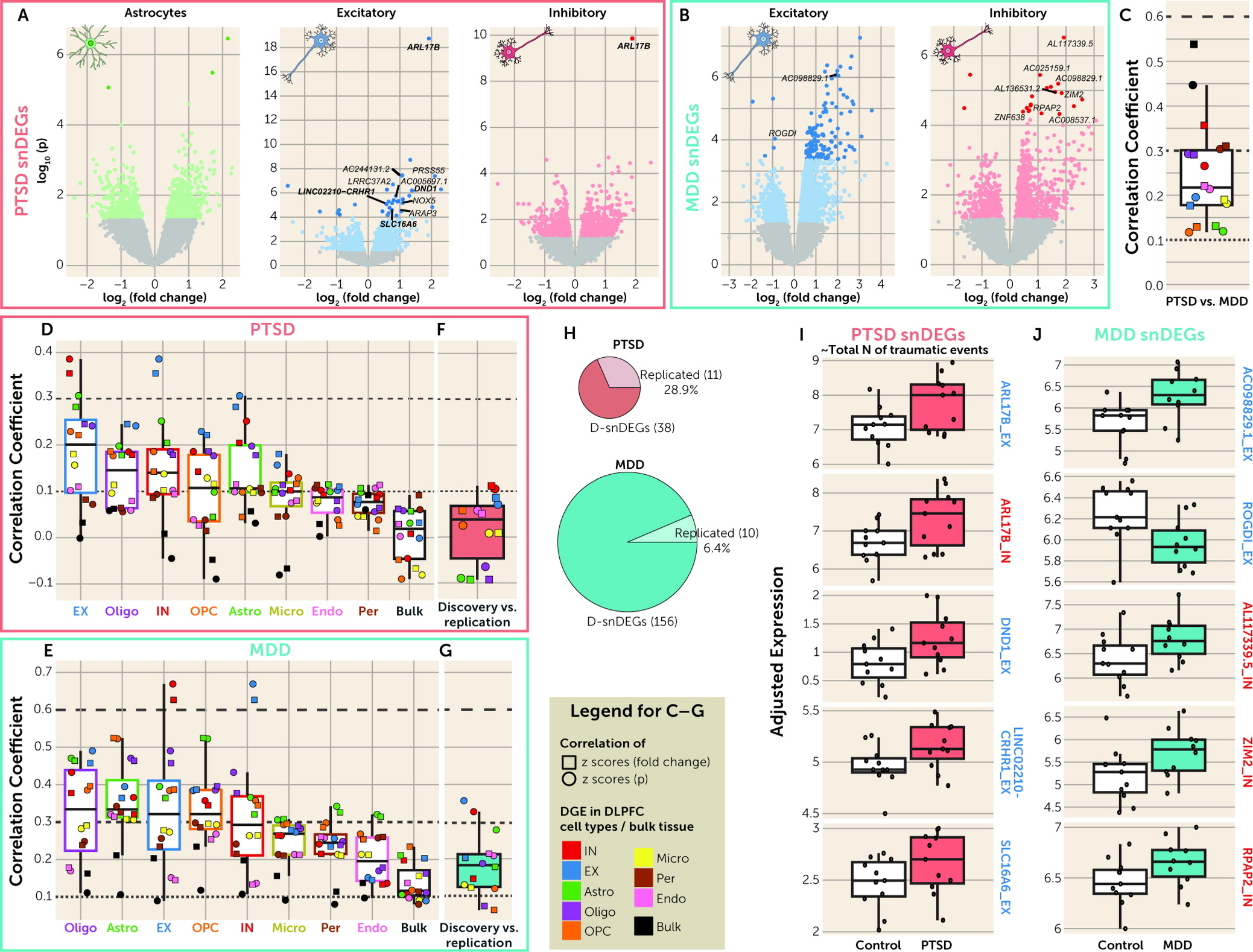
This is the first single-nucleus RNA-seq study that dissects cell-type-specific transcriptomic alterations in PTSD. We profiled ∼415K single nuclei belonging to eight cell types from postmortem samples of individuals with PTSD, individuals with MDD, and control subjects. The disorders were similar in cell type proportions and differed in specific disease-associated transcriptomic alterations in neurons and astrocytes, observations replicated in an independent data set (N=15 per group, ∼160K single nuclei). Among these different expression patterns in PTSD compared with MDD, we associated distinct biological pathways in neuronal cell types, including the GC pathway. We further compared the expression signatures of PTSD and MDD in EX and IN neurons with GR-dependent GC-induced DEGs of iPSC-derived neurons and demonstrated opposite directions of enrichment between the two disorders. To prioritize top genes for PTSD and MDD, we generated sets of putative causal genes based on SMR and/or TWAS analyses for PTSD and MDD, and TWAS analysis of DLPFC-based neuroimaging.
This study, along with a schizophrenia study in preprint (
28), profiled significantly more cells (∼500K) compared with earlier DLPFC single-nucleus RNA-seq studies in ASD (∼100K [
25]) and MDD (∼80K [
26]). Using similar technical and bioinformatic pipelines, we confirmed consistent cell subtype annotations. It is worth noting that our study was concordant with the astrocyte subtyping of the ASD study (
25) and agreed on the development-based pseudo time distinctions between OPC and oligodendrocyte cell clusters of the MDD study (
26).
While large-scale, deeply sequenced bulk-tissue RNA-seq study designs can provide substantial insight into the pathophysiology of diseases, our study design showed higher discoverability of DEGs in the snRNA-seq portion of the data, compared with the bulk-tissue RNA-seq, within a relatively limited sample size. This demonstrates the benefits of single-cell transcriptomic approaches in the field of complex psychiatric disorders, where it remains to be fully exploited. PTSD and MDD D-snDEGs were found almost entirely in EX and IN neurons compatible with the cell type enrichment of PTSD and MDD GWASs and bulk-tissue RNA-seq studies showing downregulation of interneuron markers in PTSD (
22–
24).
In line with the previous bulk-tissue RNA-seq studies (
22–
24), MDD had more D-snDEGs compared with PTSD. MDD had higher genome-wide correlations between the discovery and replication samples compared with PTSD, while there was a higher degree of replication of top snDEGs in PTSD compared with MDD. This was partially described in bulk-tissue RNA-seq studies, where low concordance in top DEGs was seen in MDD (
20–
22) and a higher level of concordance in top DEGs in PTSD (
22,
23). Our study and the prior MDD single-nucleus RNA-seq study agreed on the reduced GR activity in MDD (
26). We did not detect FDR-significant effects in OPCs as seen previously (
26), but we showed a significant enrichment of MDD GWAS in OPC markers. It is worth noting that the prior study used a male cohort in which all MDD cases were suicide completers (only 20% of the subjects of the present study were suicide completers), conducted subclustering of OPC/oligodendrocyte cell clusters, and analyzed only highly expressed genes for differential expression between groups.
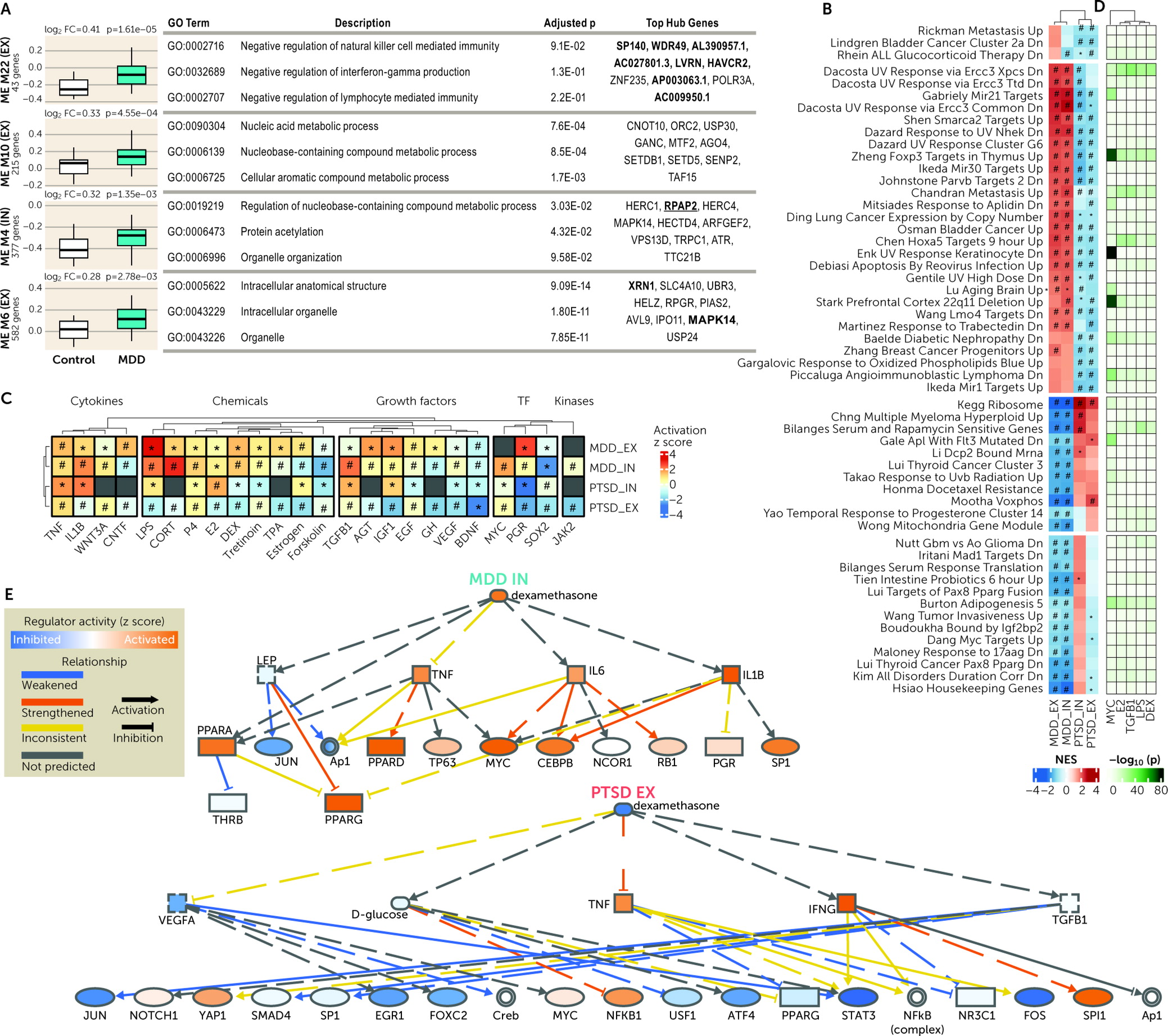
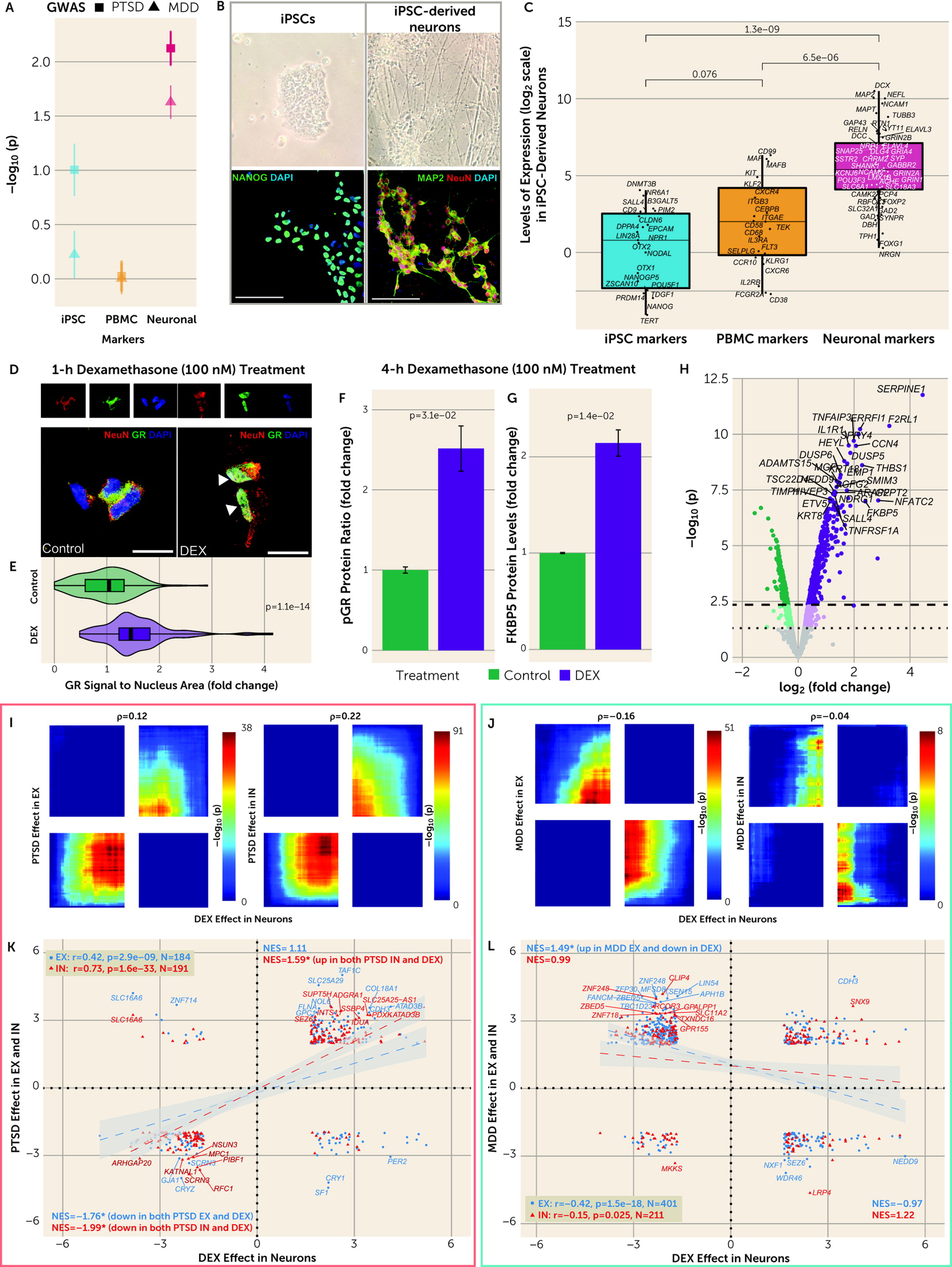
There were multiple dysregulated pathways in both EX and IN neurons across disorders. Importantly, there were pathways with a differential enrichment in PTSD compared with MDD. The involved pathways included immunologic, cancer-related, metabolic, and various ribosomal and enzymatic processes. Predicted upstream regulators tracked together with the cell-type-specific pathways. For instance, the GC treatment pathway differentiated the two traits, while GCs were also predicted to be upstream regulators. Moreover, neuronal GC-induced transcriptomic alterations were studied in iPSC-derived neuronal cultures, and these converged with gene set enrichment analyses and genome-wide correlations on DLPFC EX and IN neurons’ transcriptional signatures in PTSD and MDD in different directions. These transcriptional patterns showed GR activation in EX and IN neurons in PTSD and GR deactivation in EX neurons in MDD, with the latter being generally weaker. These results are the first molecular demonstration of cell-type-specific directional differences in GC pathways between PTSD and MDD in a stress-related brain region. These differences are consistent with differences historically described on peripheral endocrine measures of the HPA axis and PBMC-based GC sensitivity assays (
8,
30). Due to the maturity of our in vitro models, we observed
FKBP5 and genome-wide transcriptomic responses to GC that overlapped with those described in iPSC-derived cerebral organoids (
36) and were greater than those described in less mature iPSC-derived neural cells from embryoid body-based (
37) and NGN2-based (
38) neuralization protocols. Conceptually, our DEX-treated iPSC-derived neuron cultures represent an in vitro neuronal model optimized for studying a brain-based risk-associated pathway of PTSD. Future in vitro modeling studies in PTSD can examine GC responses in additional brain cell types and in novel cell lines that recapitulate PTSD’s genetic and epigenetic pathology, as well as further clarify whether they are mediated by GR and/or MR.
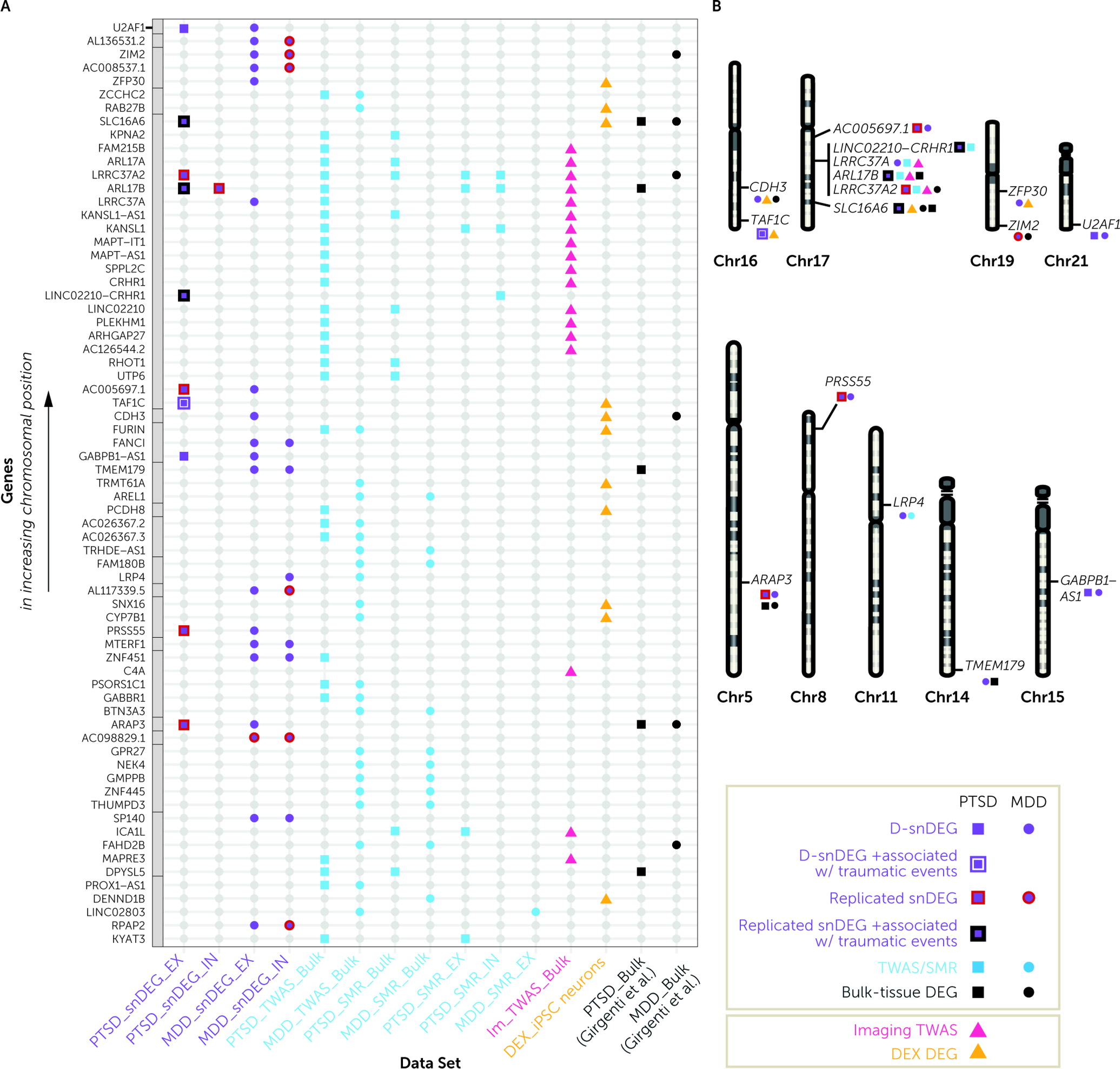
To determine the genes with a key role in disease states, we prioritized snDEGs that overlapped with bulk-tissue and/or cell-type-specific analyses of the largest GWASs of PTSD, MDD, and neuroimaging. To determine causation of genes for PTSD and/or MDD, we used SMR analysis on both bulk and cell-type-specific eQTL DLPFC databases (
33,
34). To link the combination of genetic risk variants with changes in expression, we used TWAS. We trained a state-of-the-art DLPFC GReX model that was more accurate and predicted more genes than previous models generated by the GTEx project (
39). We also revealed genes with preferential TWAS signals for DLPFC-based neuroimaging in UK Biobank (N∼33K).
The observed overlap between snDEGs and genes discovered by the genetic analyses underscored the functional involvement of genes at the 17q21.31 locus, which are already implicated in neurodevelopmental (
40) and neurodegenerative disorders (
41).
ARL17B was upregulated in PTSD across bulk-tissue, neuronal, and nonneuronal analyses and encodes for a GTP-binding protein involved in protein trafficking.
ARL17B is upregulated in multiple cell types in carriers of a missense variant of
SPPL2C associated with onset age in Alzheimer’s disease (
42).
LINC02210-CRHR1 was upregulated in EX neurons and is a fusion gene between
LINC02210 and
CRHR1.
LINC02210 and
CRHR1 were not differentially expressed in the postmortem data but were putative causal genes for PTSD and had effects on DLPFC-based neuroimaging.
CRHR1 encodes the CRHR1 protein, a master regulator of the stress and fear response that has long been thought to be involved in PTSD and MDD.
LINC02210 encodes a long noncoding RNA with a relatively unknown function, emphasizing the emerging role of noncoding RNAs in the pathophysiology of stress-related mental disorders (
43). Long noncoding RNAs act as regulators of transcription, affecting many downstream target genes. Given associations of
LINC02210 with DLPFC-based neuroimaging, future studies should expand the comparisons of coding versus noncoding genetic associations with neuroimaging. For instance, it would be very interesting to understand whether cell subtypes with dysregulation in noncoding genes are in a particular spatial brain compartment that would justify more influences in white matter specifically.
LRRC37A2 was upregulated in PTSD EX neurons and encodes an immune-related leucine-rich repeat protein involved in the formation of protein-protein interactions.
LRRC37A, a paralog of
LRRC37A2, was an upregulated bulk-tissue TWAS gene for PTSD, while it was also downregulated in MDD EX neurons, indicating directional differences between PTSD and MDD in this genomic region.
Outside of the 17q21.31 locus,
LRP4 was downregulated in MDD IN neurons and encodes for a member of the lipoprotein receptor-related protein family with known roles in neurodevelopment, synaptic remodeling, and Alzheimer’s disease (
44). Furthermore, the upregulation in PTSD of the GC-regulated
SLC16A6 in EX neurons might be a link to mitochondrial regulation of physiological and behavioral responses to psychological stress (
45). SLC16A6 belongs to the monocarboxylate transporter (MCT) family that contributes to maintenance of intracellular pH; MCT7/SLC16A6 is expressed in the liver, brain, and endocrine tissues and is classified as a transporter for ketone bodies during mitochondrial ketogenesis. As such, it is expected to have a multitude of physiological roles (
46).
CDH3 was upregulated in MDD EX neurons and was induced by DEX in iPSC-derived neurons.
CDH3 encodes a P-cadherin that has been associated with neuronal development and differentiation (
47). The
TAF1C gene was upregulated in PTSD IN neurons and by DEX in iPSC-derived neurons. This upregulation could result in proteomic alterations, since
TAF1C encodes the largest subunit of the SL1 complex, which is required for RNA polymerase I to bind to promoters of ribosomal RNA genes (
48).
A limitation of this single-nucleus RNA-seq study is the sample size of the discovery data set. We attempted to mitigate this limitation by profiling a high number of cells, using a replication data set, seeking biological validation of discovered pathways, convergence with putative causal genes, and assessing relevance to DLPFC-based neuroimaging. While these efforts contributed to the robustness of our findings, further validation is needed. Another limitation of postmortem analysis of PTSD is that while in-depth clinical characteristics of the PTSD collection were assessed systematically (
49), assessments were based on interviews with next of kin and medical record review. We also acknowledge that the replication sample was also small and did not fully match our discovery sample. These demographic differences strengthened our observed levels of replication. Our data sets included only African and European ancestries, however, and generalizations to other populations warrant careful consideration. Finally, the evaluation of trauma exposure–based effects is a novel approach in postmortem research. Recent postmortem studies investigating trauma-based subgroup approaches across psychiatric disorders provide a level of precedence and confidence (
50), although these approaches need to be further replicated in larger samples.
From a technical perspective, studying the gray versus white matter contributions to cell-type-specific transcriptomic disease signatures may be critical for revealing nonneuronal signals. To this end, single-nucleus RNA-seq may not adequately sample perivascular components, i.e., pericytes (e.g., detected in our discovery data set but not the replication data set). Future studies could use more refined techniques, such as vessel isolation and nucleus extraction for sequencing (VINE-seq), to enrich vascular and perivascular nuclei to profile endothelial cells and pericytes more comprehensively (
51). Additionally, single-nucleus RNA-seq introduces some level of bias in the detection of certain gene types, as nonpolyadenylated transcripts cannot be detected (including ribosomal RNAs, small RNAs, histone mRNAs, and some long noncoding RNAs (
52). As described by Thrupp et al. (
53), a small but nonnegligible proportion of microglia genes (∼1.1%) are not detected by single-nucleus RNA-seq, which can lead to underrepresentation of certain cellular states.
In summary, in this first single-cell transcriptomic investigation of PTSD, the comparison of PTSD with MDD results enabled us to identify shared and unique neuronal genes and targetable stress-related gene pathways, which we validated with in vitro modeling. The overlap of stress-related gene pathways with disease causal genes and specific brain connectivity measures provides an avenue for future translational investigation.
Acknowledgments
The authors acknowledge Kiki Galani, Bob Handsaker, Li-Lun Ho, Maria Kousi, Shahin Mohammadi, and Shreejoy Tripathy for suggestions in the choice of methods, technical assistance, and/or presentation of data. The authors acknowledge three working groups: The members of the Traumatic Stress Brain Research Group are Matthew Friedman, Neil Kowall, Christopher Brady, Ann McKee, Thor Stein, Bertrand Huber, Paul Holtzheimer, Victor Alvarez, David Benedek, Robert Ursano, Douglas Williamson, Dianne Cruz, Keith Young, John Krystal, Deborah Mash, Melanie Hardegree, William Scott, David Davis, Matthew Girgenti, Gayle Serlin, Brian Marx, Terence Keane, Mark Logue, Erika Wolf, and Mark Miller. The members of the PTSD BrainOmics Project of the PsychENCODE Consortium are Dhivya Arasappan, Sabina Berretta, Rahul Bharadwaj, Frances Champagne, Leonardo Collado-Torres, Christos Chatzinakos, Nikolaos Daskalakis, Chris DiPietro, Duc Duong, Amy Deep-Soboslay, Nick Eagles, Louise Huuki, Thomas Hyde, Artemis Iatrou, Aarti Jajoo, Joel Kleinman, Charles Nemeroff, Geo Pertea, Deanna Ross, Nicholas Seyfried, Joo Heon Shin, Kerry Ressler, Clara Snijders, Ran Tao, Daniel Weinberger, Stefan Wuchty, Dennis Wylie. The members of the PTSD Working Group of the Psychiatric Genomics Consortium are Adam X. Maihofer, Karmel W. Choi, Jonathan R.I. Coleman, Nikolaos P. Daskalakis, Christy A. Denckla, Elizabeth Ketema, Rajendra A. Morey, Renato Polimanti, Andrew Ratanatharathorn, Katy Torres, Aliza P. Wingo, Clement C. Zai, Allison E. Aiello, Lynn M. Almli, Ananda B. Amstadter, Soren B. Andersen, Ole A. Andreassen, Paul A. Arbisi, Allison E. Ashley-Koch, S. Bryn Austin, Esmina Avdibegovic, Anders D. Borglum, Dragan Babic, Marie Bækvad-Hansen, Dewleen G. Baker, Jean C. Beckham, Laura J. Bierut, Jonathan I. Bisson, Marco P. Boks, Elizabeth A. Bolger, Bekh Bradley, Meghan Brashear, Gerome Breen, Richard A. Bryant, Angela C. Bustamante, Jonas Bybjerg-Grauholm, Joseph R. Calabrese, J.M. Caldas-de-Almeida, Chia-Yen Chen, Anders M. Dale, Shareefa Dalvie, Jürgen Deckert, Douglas L. Delahanty, Michelle F. Dennis, Seth G. Disner, Katharina Domschke, Laramie E. Duncan, Alma Dzubur Kulenovic, Christopher R. Erbes, Alexandra Evans, Lindsay A. Farrer, Norah C. Feeny, Janine D. Flory, David Forbes, Carol E. Franz, Sandro Galea, Melanie E. Garrett, Aarti Gautam, Bizu Gelaye, Joel Gelernter, Elbert Geuze, Charles F. Gillespie, Aferdita Goci, Scott D. Gordon, Guia Guffanti, Rasha Hammamieh, Michael A. Hauser, Andrew C. Heath, Sian M.J. Hemmings, David Michael Hougaard, Miro Jakovljevic, Marti Jett, Eric Otto Johnson, Ian Jones, Tanja Jovanovic, Xue-Jun Qin, Karen-Inge Karstoft, Milissa L. Kaufman, Ronald C. Kessler, Alaptagin Khan, Nathan A. Kimbrel, Anthony P. King, Nastassja Koen, Henry R. Kranzler, William S. Kremen, Bruce R. Lawford, Lauren A.M. Lebois, Catrin Lewis, Israel Liberzon, Sarah D. Linnstaedt, Mark W. Logue, Adriana Lori, Bozo Lugonja, Jurjen J. Luykx, Michael J. Lyons, Jessica L. Maples-Keller, Charles Marmar, Nicholas G. Martin, D. Maurer, Matig R. Mavissakalian, Alexander McFarlane, Regina E. McGlinchey, Katie A. McLaughlin, Samuel A. McLean, Divya Mehta, Rebecca Mellor, Vasiliki Michopoulos, William Milberg, Mark W. Miller, Charles Phillip Morris, Ole Mors, P.B. Mortensen, Elliot C. Nelson, Merete Nordentoft, Sonya B. Norman, Meaghan O’Donnell, Holly K. Orcutt, Matthew S. Panizzon, Edward S. Peters, Alan L. Peterson, Matthew Peverill, Robert H. Pietrzak, Melissa A. Polusny, John P. Rice, Victoria B. Risbrough, Andrea L. Roberts, Alex O. Rothbaum, Barbara O. Rothbaum, P. Roy-Byrne, Kenneth J. Ruggiero, Ariane Rung, Bart P.F. Rutten, Nancy L. Saccone, Sixto E. Sanchez, Dick Schijven, S. Seedat, Antonia V. Seligowski, Julia S. Seng, Christina M. Sheerin, Derrick Silove, Alicia K. Smith, Jordan W. Smoller, Scott R. Sponheim, Dan J. Stein, Jennifer S. Stevens, Martin H. Teicher, Wesley K. Thompson, Edward Trapido, Monica Uddin, Robert J. Ursano, Leigh Luella van den Heuvel, Miranda Van Hooff, Eric Vermetten, Christiaan Vinkers, Joanne Voisey, Yunpeng Wang, Zhewu Wang, Thomas Werge, Michelle A. Williams, Douglas E. Williamson, Sherry Winternitz, Christiane Wolf, Erika J. Wolf, Rachel Yehuda, Keith A. Young, Ross McD. Young, Hongyu Zhao, Lori A. Zoellner, Magali Haas, Heather Lasseter, Allison C. Provost, Rany M. Salem, Jonathan Sebat, Richard A. Shaffer, Tianying Wu, Stephan Ripke, Mark J. Daly, Kerry J. Ressler, Karestan C. Koenen, Murray B. Stein, and Caroline M. Nievergelt.