Huntington's disease (HD) is an autosomal inherited neurodegenerative disorder characterized by choreic movements and disturbed voluntary movements, personality changes, and a general cognitive decline.
1 The mean age at onset is 40 years (range 2–80 years). The mean duration of the illness is 17 years (range 2–43 years). In 1983, the gene was determined to be located on chromosome 4,
2 and 10 years later the mutation was identified as a number of CAG repeats exceeding 35.
3 This allowed gene carriers to be determined, with 99% certainty, prior to onset of signs of HD.
Because of the great variety of symptoms and because signs of HD develop gradually, it is difficult to determine the precise clinical onset of the disease. The diagnosis of HD was usually based on family history and the appearance of motor (i.e., choreic) symptoms. However, cognitive impairment, motor symptoms, and behavioral and emotional changes differ in time of onset and severity. Genetic testing for prolonged CAG repeat offers a unique opportunity for studying the beginning and progression of the disease by following the group of at-risk individuals who underwent predictive testing. Recognition of early signs seems important in view of potential future neuroprotective treatments. An extended follow-up is required to gain more insight into the clinical manifestations of gene carriers.
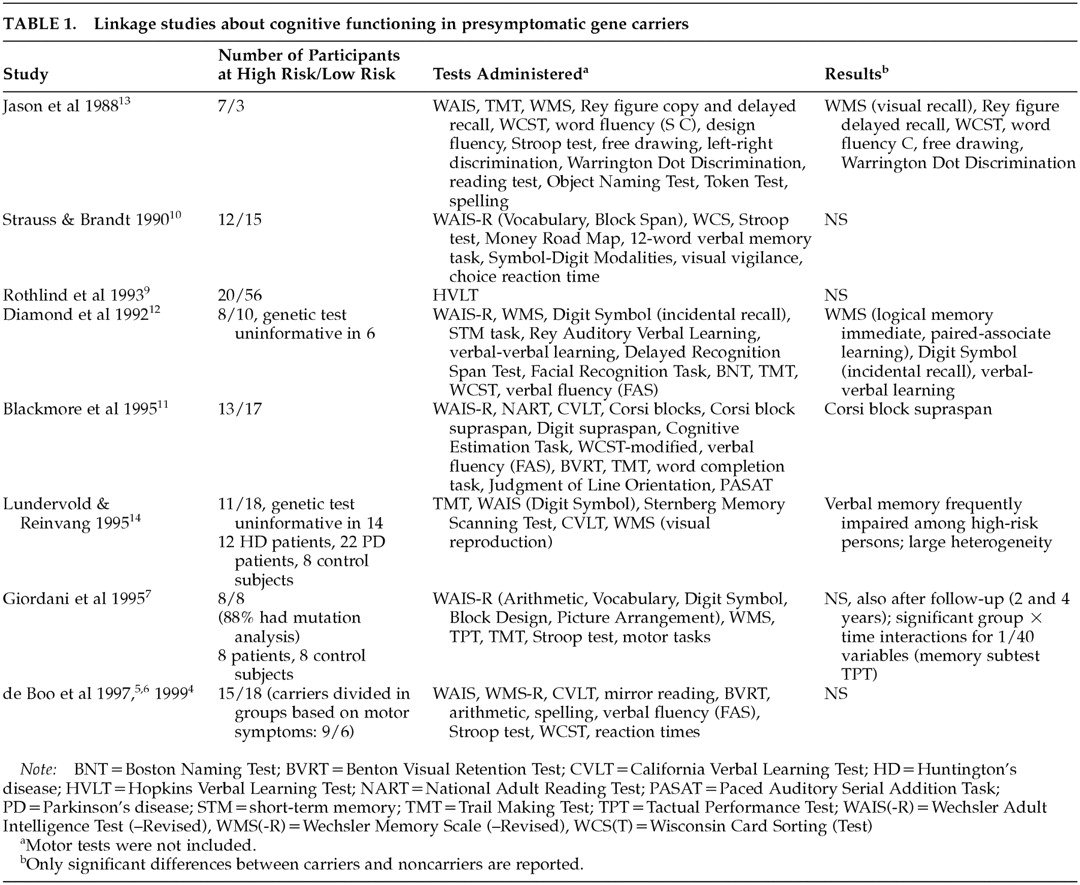
Most studies on cognitive functioning using linkage analysis to determine carriership show no differences between gene carriers and non–gene carriers prior to symptom onset,
4–10 but a few have found certain changes in the gene carrier group
11–14 (
Table 1).
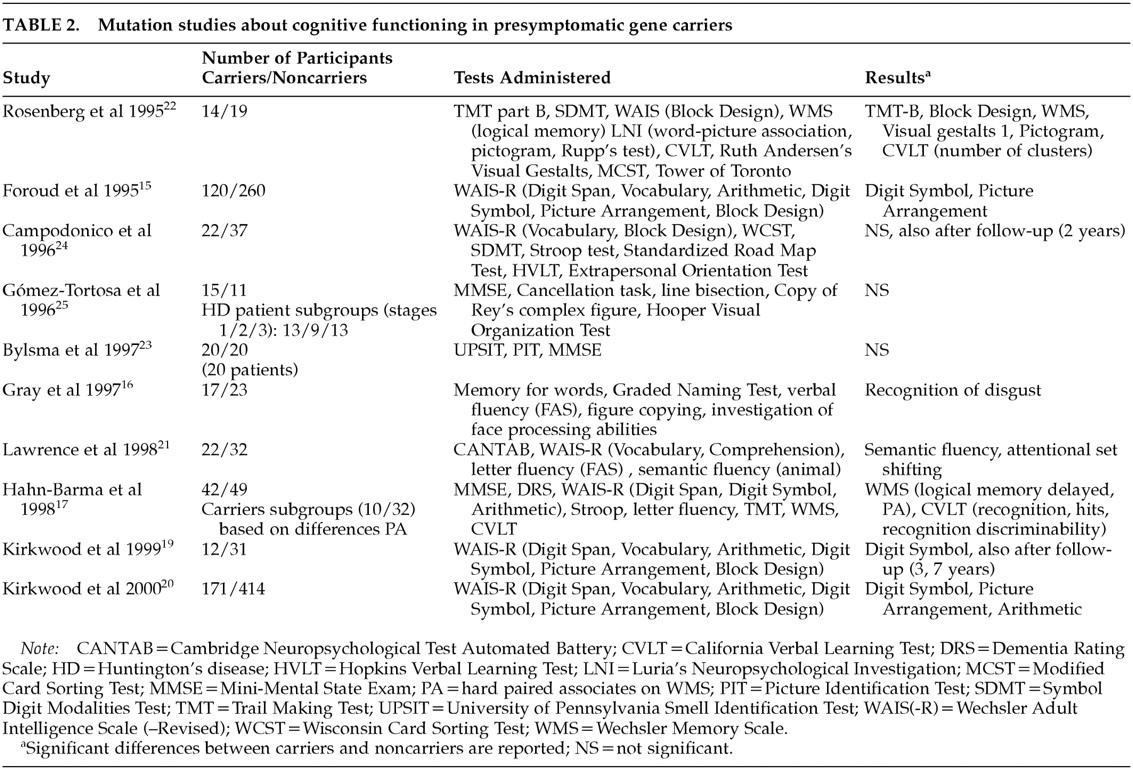
In studies involving mutation analysis, results are also equivocal and contradictory. Some suggest that subtle cognitive impairment is present before the appearance of manifest motor symptoms of HD,
15–22 while others refute this finding.
23–25 Table 2 shows the findings of these studies about cognitive functioning in gene carriers compared with non–gene carriers.
With the exception of the studies by Foroud et al.,
15 Hahn-Barma et al.,
17 and Kirkwood et al.,
20 the number of participants was small. All studies focused on a limited number of cognitive domains. So far, only two follow-up studies have been reported.
19,24 The relationship between the number of CAG repeats and cognitive functioning has also been the subject of investigation. A lower performance associated with a longer repeat has been suggested by cross-sectional studies.
15,17,18,20Because the onset of the clinical manifestations remains unclear and few follow-up studies have been undertaken with this particular group, the present study was set up to compare presymptomatic gene carriers of Huntington's disease with non–gene carriers with respect to cognitive and motor functioning. An earlier pilot study with a comparable design was performed at our department with participants who underwent linkage analysis.
26 Unfortunately, this group was too small for follow-up because of insufficient response.
METHODS
Participants
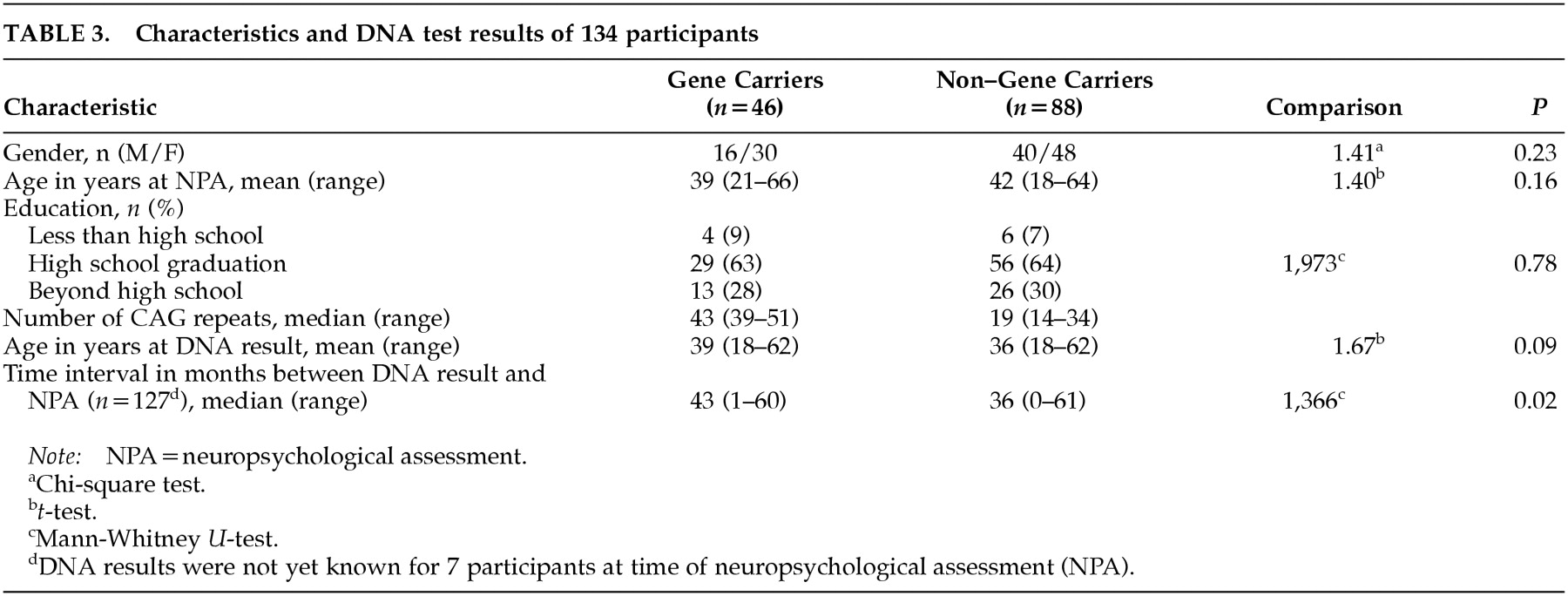
Between 1993 and the end of 1998, 370 persons at 50% risk for Huntington's disease came to the Leiden University Medical Center (LUMC) for presymptomatic counseling by direct mutation testing. Predictive testing was performed according to the guidelines of the International Huntington Association and the World Federation of Neurology, Research Group on Huntington Disease.
27 Participants were considered HD gene carriers when the number of CAG copies exceeded 35 repeats. Participants with a repeat containing fewer than 27 copies were considered to be non–gene carriers. Those with a repeat number between 27 and 35 were considered intermediate.
28All 370 applicants were invited by mail to participate in this study. Gene carriers who were already affected at the time of the DNA testing procedure were excluded. Between November 1997 and January 1999, 134 individuals (36%) participated in the initial assessment of this single-blind study. They included 12 participants who had previously undergone the linkage test and received direct testing after 1993. For seven participants, the study was double-blind because they did not yet know the outcome of their DNA test at entry. Three participants had an intermediate result (CAG repeats: 30, 30, 34); for the present study they were included in the noncarriers group because they are unlikely to develop the disease.
28The study was approved by the Medical Ethics Committee from the LUMC, and all participants gave their informed consent.
Procedure
All participants were explicitly asked not to disclose the result of DNA analysis to ensure that the neuropsychological investigators could carry out a blind assessment.
Systematic history was taken by a psychological assistant, and all participants were asked if they had any complaints about their functioning at that point in time. The questions concerning complaints covered the following categories: memory, concentration, motor, affect, behavior, and others (mainly headache, backache, and fatigue).
Furthermore, all participants were assessed with the Unified Huntington's Disease Rating Scale (UHDRS),
29 consisting of questions about medical history, a motor examination, a behavioral assessment, an assessment of functional ability, and a medication form. The motor part of the UHDRS was blindly scored by a neurologist (J.P.P.v.V. or R.A.C.R.) who had no information about any of the participants. Diagnosis based on this part of the assessment was classified into four categories: normal, “minor soft signs,” probable HD, and unequivocal HD.
The Mini-Mental State Examination
30 was administered, along with a broad neuropsychological assessment of tests established to investigate cognitive functioning (
Appendix A): intelligence, memory and learning abilities, language skills, calculation skills, visuoconstructive skills, mental and motor speed, and the so-called executive functions.
The choice of the test battery was based on tests used in previous studies involving patients with HD and on the CAPIT-HD protocol used by the Huntington Research Group.
31 Standard instructions were given at the beginning of each test. The full assessment was administered over a period of 4 to 5 hours in a strictly prescribed order (including lunch break).
The rating of cognitive impairment was determined by using the discrepancy between the estimated premorbid IQ, based on education, profession, and intact abilities, and the current IQ and the Memory Quotient of the Wechsler Memory Scale. Impairment was classified into four categories: none (discrepancy<10 points), mild (11–15 points), moderate (16–25 points), and severe (>25 points). In addition, a qualitative judgment was made concerning impairment of specific disorders in the areas of calculation, language, and constructional abilities, which was done on the basis of the scores achieved or on performance levels in the tests.
32Statistical Analyses
All test results were scored on special forms, and DNA test results were encoded before being processed with the SPSS statistical software package. Data were analyzed with two-tailed parametric (Student's t-test) and nonparametric statistics (Pearson chi-square test and Mann-Whitney U-test). If the number of participants in a category was too small (n<10), Fisher's exact test was reported. Logistic regression analyses were also performed.
Because of the large number of comparisons, the Bonferroni correction was applied to set a significance level of alpha=0.001.
33 A more liberal
P level (<0.01) is also reported.
RESULTS
The percentage of gene carriers among study participants (34%;
n=46) was lower compared with the whole group of applicants for DNA testing (44%). The difference was marginally significant (
P=0.006). No significant differences were found between participants and nonparticipants when comparing for gender (χ
2=0.035, df=1,
P=0.85) and age (participants, mean=40.7, SD=11.3; nonparticipants, mean=38.6, SD=11.8;
t=–1.69, df=368,
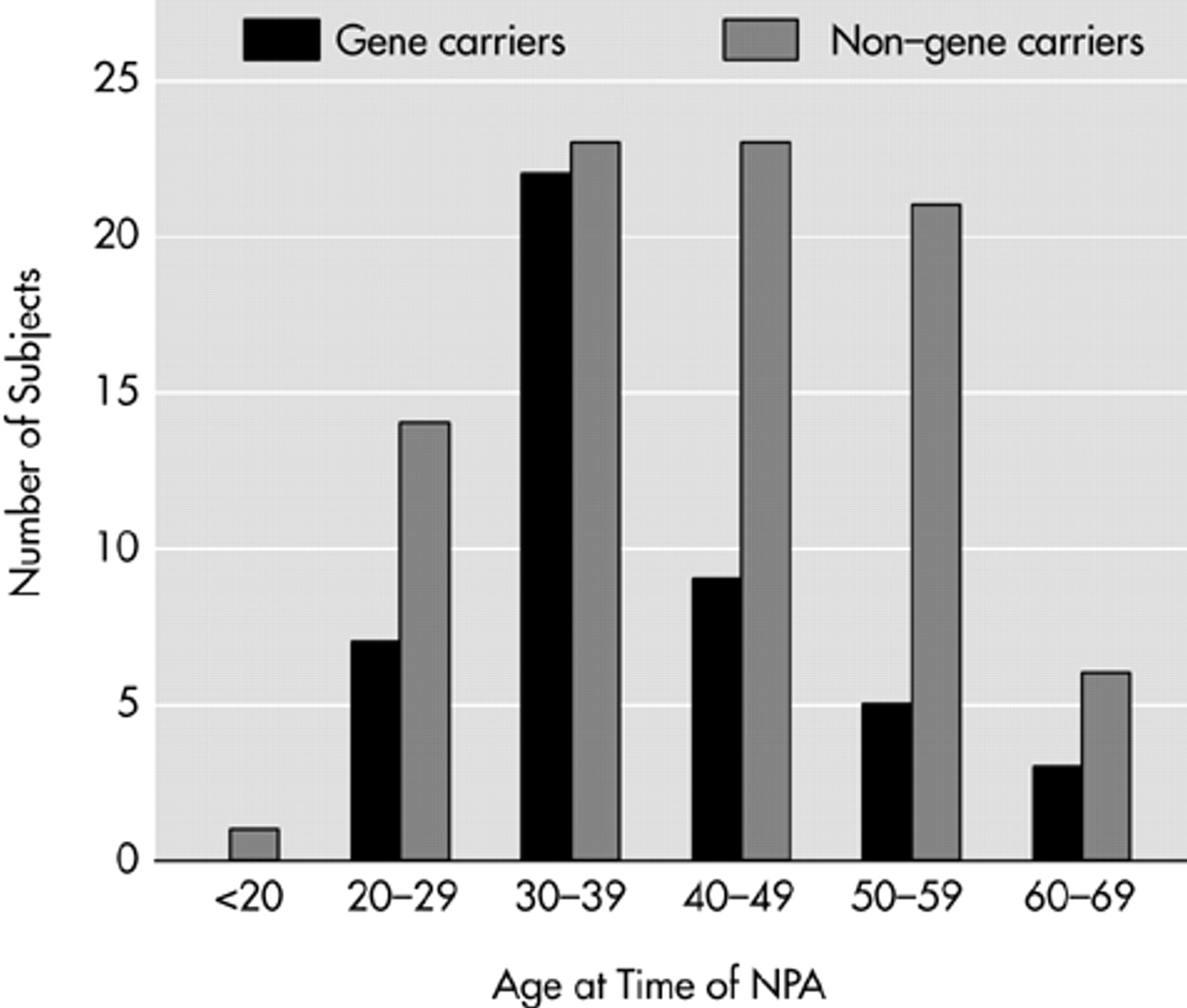
P=0.09). However, among the participants, half of the non–gene carriers were between 40 and 60 years old compared with a smaller percentage in the gene carrier participants (
Figure 1). The reverse pattern was found in the nonparticipants' group. The demographic characteristics of the participants are presented in
Table 3.
The groups did not differ in the number or type of complaints about their functioning (Mann-Whitney U=2,001, P=0.90). Less than 10% of the gene carriers complained about memory, concentration, motor symptoms, and behavior symptoms; 15% complained about their affect; and 24% had “other” complaints. Percentages of complaints within non–gene carriers were 15% for memory, 5% for affect, and 32% for “other,” but the difference from gene carriers was not statistically significant.
The classification of the two groups based on the UHDRS motor assessment is presented in
Table 4. “Minor soft signs” were rated equally often in both groups. Fifteen participants were classified according to the UHDRS result as having either probable or unequivocal HD, but the percentage was twice as high in the group of gene carriers compared with the non–gene carriers.
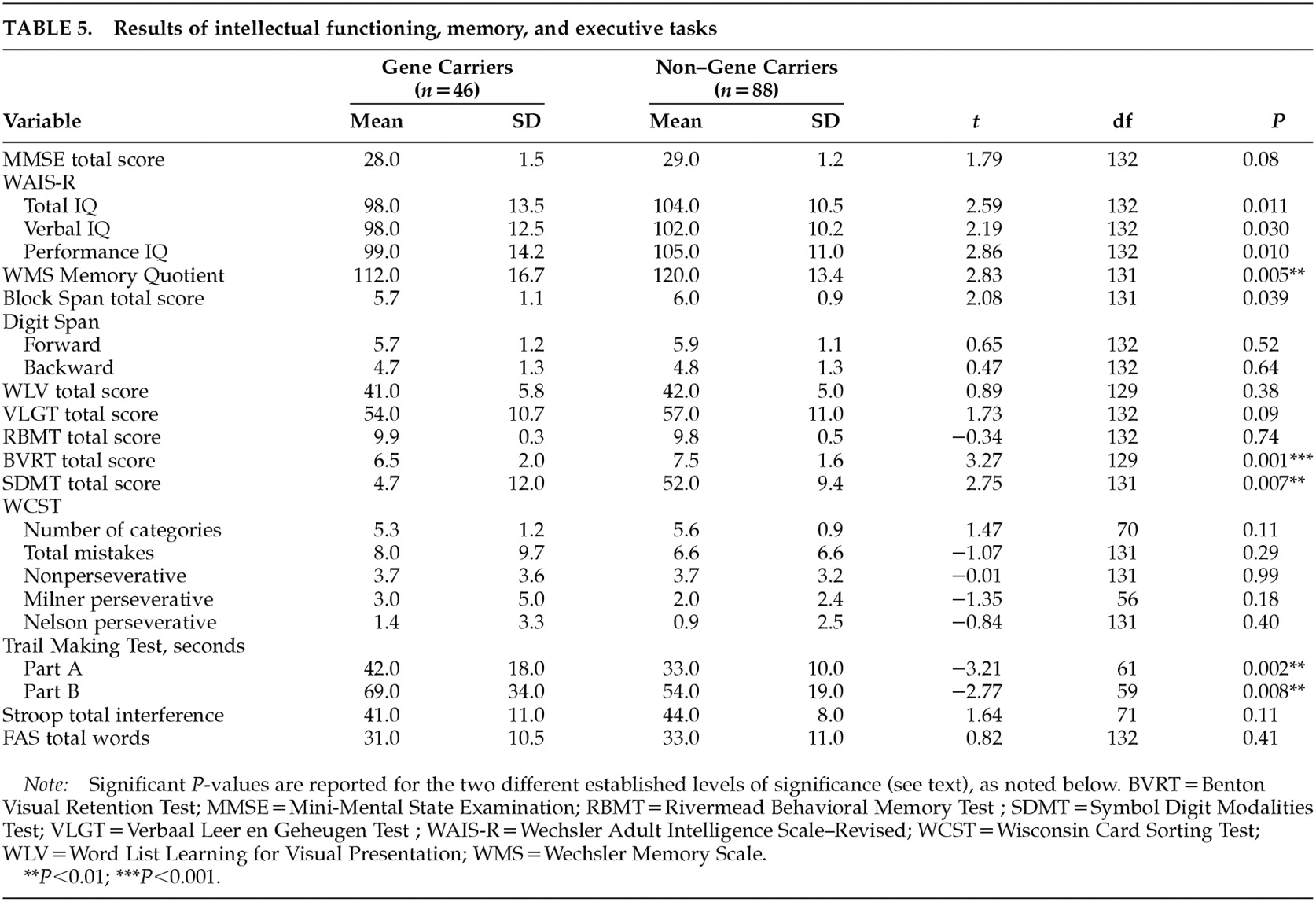
The results of intellectual functioning, memory, and executive tasks are presented in
Table 5. Gene carriers performed significantly worse on the Benton Visual Retention Test (
P=0.001). This result changed slightly after correction for age and total IQ (
P=0.002). A
P-value of less than 0.01 was found on the Memory Quotient, the Symbol Digit Modalities Test, and the Trail Making Test parts A and B. Reaction time measures showed only a marginal significance in the motor time from the choice light/light condition (gene carriers, mean=180, SD=61; non–gene carriers, mean=149, SD=44;
t=–3.21, df=124,
P=0.002). None of the other tests in the battery had significantly different results. Within the visuoconstructive tasks, three gene carriers (6%) were rated as disturbed in figure drawing; the one rated as “seriously disturbed” was also judged to have global changes in cognitive functioning. Unsteady line drawing was seen in 9% of the gene carriers and in 10% of the non–gene carriers.
When we looked at the rating of cognitive impairment, four gene carriers (number of CAG repeats: 44–47) and one non–gene carrier (number of CAG repeats: 18) were found to be impaired (four mildly and one moderately). Without these five participants, the significant difference between the two groups on the Benton Visual Retention Test was reduced (gene carriers, mean=6.6, SD=2; non–gene carriers, mean=7.5, SD=1.6; t=2.73, df=125, P=0.007), and the P-levels for the other tests were all greater than 0.01 with the exception of the Trail Making Test part A (gene carriers, mean=38.1, SD=13; non–gene carriers, mean=32.4, SD=10.4; t=–2.66, df=127, P=0.009) and the motor time light/light measure (gene carriers, mean=176.7, SD=59.4; non–gene carriers, mean=149.2, SD=44.4; t=–2.84, df=120, P=0.005).
Analyses were also carried out excluding the 15 participants rated as having probable or unequivocal HD according to the UHDRS. The number of the CAG repeats for this group varied between 39 and 49 in the gene carriers and between 16 and 26 in the non–gene carriers. The differences on the above-described tests disappeared, but marginal differences were found between gene carriers and non–gene carriers on three motor times of reaction time measures (single light: gene carriers, mean=191.7, SD=64.9; non–gene carriers, mean=160, SD=47.2; t=–2.80, df=102, P=0.006; single sound: gene carriers, mean=171.2, SD=65.9; non–gene carriers, mean=139.8, SD=41.2; t=–2.94, df=101, P=0.004; choice light/light: gene carriers, mean=183.7, SD=67; non–gene carriers, mean=144.5, SD=43.6; t=–3.03, df=43.09, P=0.004).
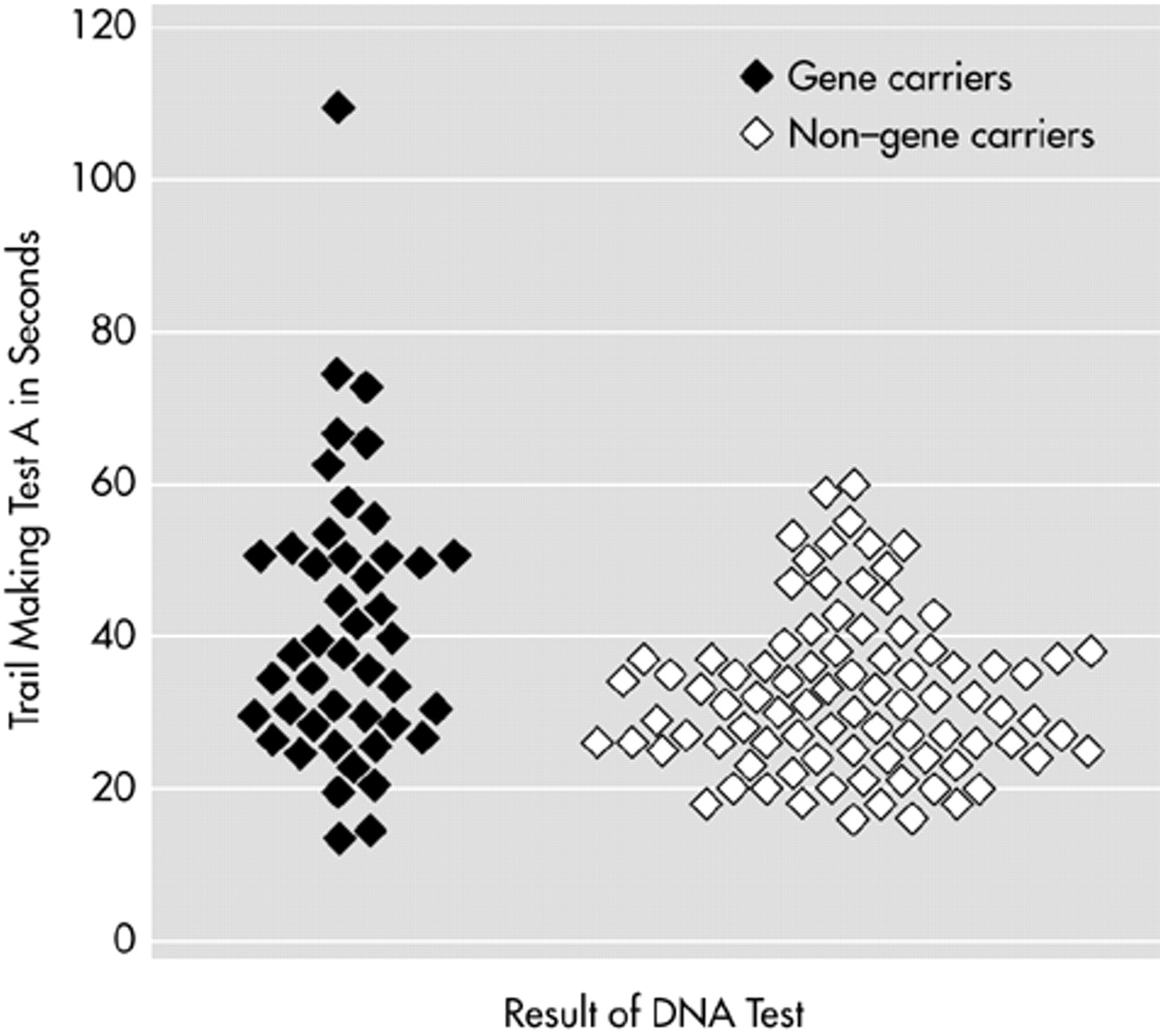
Stepwise logistic regression analysis was performed on the total group to investigate which test could best discriminate between gene carriers and non–gene carriers. Trail Making A was selected as the best indicator for gene carrier status (
P<0.001), but it explained only 7% of the variance (
Figure 2).
DISCUSSION
Diagnosis of a person as symptomatic for HD is still typically based on the apparent motor changes. In this study we investigated whether gene carriers who have no clinical HD diagnosis prior to participation already present cognitive changes. We found that gene carriers did not differ from non–gene carriers, either in complaints or in the results of the neurological investigation.
Subtle motor changes in gene carriers have been demonstrated by some authors.
5,19,20,34 In our study, 60% of the gene carriers were judged to be free of motor signs, and within both groups the same percentage of participants (21%) was rated as having minor soft signs for HD. The influence of bias in rating is inevitable, since the neurologist knows that the participants are at risk of HD. Whereas substantial agreement has been demonstrated on the rating of unequivocal HD,
35 the difficulty in making an accurate diagnosis based on subtle signs of HD persists.
36 Furthermore, within neuropsychological assessment, too much overlap was found on the Trail Making Test A, although it proved to be the best differentiation of all the tests.
Differences in motor times found between gene carriers and non–gene carriers after excluding participants rated as having probable or unequivocal HD were marginal, which was unexpected. These results actually confirm the findings of de Boo et al.
36,37 with regard to the difficulty of assessing motor changes by neurological investigation. They suggest a more objective assessment by using computerized tasks, which can be a valuable addition to diagnostic methods.
Even though some results showed cognitive differences between gene carriers and non–gene carriers, our study could not determine whether cognitive changes occurred within the gene carriers as a group. A small number of gene carriers (ages: 32, 43, 52, and 59 years) were rated as being cognitively impaired and had probably developed cognitive decline within the 3.5- to 4.5-year interval between predictive testing and entry to this study. Differences between the carrier and noncarrier groups seem to be due to this minor sample, because when these participants were excluded, the differences disappeared. The gene carriers of this subgroup differed in their ratings on the UHDRS: one normal, one with minor soft signs, one with unequivocal HD, one not administered. For the first two of these participants, cognitive changes may have appeared as an initial sign of HD, but this is unknown for the one also presenting clear motor signs.
An extended battery of tests was used because many domains of cognitive functioning become affected in HD
38 and because of the discrepancies in reported results so far (
Table 1 and
Table 2). Most previous studies about this subject found no differences between gene carriers and non–gene carriers on cognitive functioning.
4–10,23–25 However, two of these studies suggest that persons who are approaching clinical onset of HD may develop minor deficits in some domains such as odor identification
23 and sustained attention and mental processing speed.
24 The studies that did find differences in cognitive functioning between gene carriers and non–gene carriers showed inconsistent results (
Table 1 and
Table 2).
Are changes in cognitive functioning actually present in “presymptomatic” gene carriers for HD? Some authors are convinced they must be because even when no differences, or only trends, are found, the findings suggest that subtle deficits are present but that the tests used are not sensitive enough to reveal them.
11 Quite apart from such tests possibly being sensitive for one specific disease, their specificity is unclear because of differences in using and interpreting the various tests and the influence of many factors and cognitive domains that simultaneously play a role in test performance. Moreover, the influence of a motor component in most cognitive tasks may also play a critical role in the group studied.
Some authors suggest cognitive decline occurs prior to motor changes,
12,13,15,17,18 while others refute this contention.
4–7,10 Jason et al.
18 found similar progression in all areas assessed in affected subjects, but in their opinion it remains open whether specific areas of cognitive functions are affected earlier or more than other areas in at-risk persons.
In the years since mutation DNA testing became available, a number of cross-sectional studies have been carried out, looking at the relationship between cognitive and motor functioning in HD and the role of the CAG repeat length. In the present study, using an extended battery of tests in a large sample of participants, we obtained essentially negative findings. We believe that asserting or predicting cognitive decline in HD based on subtle differences found between gene carriers and non–gene carriers on some subtests is currently impossible. Changes such as intellectual and memory decline (assessed on the basis of education, profession, and intact abilities) must be present to speak of a sign of HD and to relate it to motor and behavioral changes. In accordance with Lawrence et al.,
21 we therefore do not suggest apparent cognitive dysfunction as the initial manifestation of HD.
Longitudinal studies, in which gene carriers are compared with themselves across time, are needed to give more insight into the actual beginning and the progression of HD. These kinds of studies have already been reported with other dementing conditions (mainly Alzheimer's disease). Tuokko and Frerichs
39 reviewed the neuropsychological literature addressing the concept of impaired cognitive functioning of insufficient magnitude to warrant a diagnosis of dementia. A significant proportion of persons with mild cognitive impairment progress to dementia over a 1- to 2-year interval. One of the best and most commonly identified predictors of decline to dementia is impairment on measures of memory. Memory impairment is also reported as an early cognitive feature of HD.
38 However, other studies reported that persons with subclinical cognitive impairment constitute a highly heterogeneous group
40 and that suitable criteria defining a preclinical state do not exist.
41“Preclinical syndromes” currently remain a research tool. The specific characteristics of HD compared with other dementias offer the opportunity to focus on the estimated age at onset and to investigate the correlation between eventual changes in tests over time and CAG repeat length. Our first follow-up was performed after 18 months with 85% of our participants, and the second follow-up is currently in progress.
ACKNOWLEDGMENTS
The authors are grateful to A. Krijnen, M. v.d. Heijden, P.J. Tazelaar, and F. Gronheid for their help in assessing the neuropsychological protocol; to Prof. Dr. A. Tibben and Dr. M.W. Zoeteweij for their valuable suggestions; and to B. Vollers for her help in language editing. Financial support was provided by the Netherlands Organization for Scientific Research (NWO), Grant 970-10-021. This paper was partially presented as a poster at the 6th European Meeting on Psychosocial Aspects of Genetics, Paris, October 1–3, 1998, and at the 18th International Meeting of the World Federation of Neurology Research Group on Huntington Disease, the Netherlands, August 28–September 2, 1999.