H ypocretin was first described in 1998 by De Lecea et al.
1 The name was coined from the words
hypo thalamus, referring to the anatomic location of neurotransmitter, and se
cretin, the gastric peptide with which hypocretin (Hcrt) was thought to share considerable homology. Using directional tag PCR subtraction, they discovered a hypothalamus-specific mRNA that was thought to encode the peptides Hcrt-1 and Hcrt-2. These peptides were restricted to neuronal bodies in the dorsal and lateral hypothalamus with fibers projecting to multiple targets in the hypothalamus, thalamus, and brainstem. At least one of the Hcrt peptides was found to be neuroexcitatory to hypothalamic cells but its precise function was not known. At around the same time, Sakurai et al.
2 described two novel neuropeptides that increased appetite when given to rats. They were named “orexin-A” and “orexin-B,” orexin coming from
orexis, which refers to appetite. Hypocretin and orexin are considered synonymous. In 1999, two American groups simultaneously published data linking the Hcrt peptide system with narcolepsy. Mignot’s group at Stanford University used positional cloning to identify an autosomal recessive mutation in one of the Hcrt receptor genes,
Hcrtr2, in narcoleptic canines.
3 Mutation analysis of 17 narcoleptic Doberman pinschers revealed the same insertion mutation in
Hcrtr2 ; the insertion was absent in all 36 comparison dogs tested. Ripley et al.
4 later showed that Hcrt ligand levels in the brain and cerebral spinal fluid (CSF) of Doberman pinschers were within normal range, supporting the theory that in canines, narcolepsy develops as a result of a mutation in the receptor gene. Only 2 weeks later, Masashi Yanagisawa’s group at the University of Texas Southwestern Medical Center published a report describing a narcolepsy-like phenotype in
Hcrt knockout mice.
5 Null mutation of
Hcrt produced an autosomal recessive phenotype remarkably similar to human narcolepsy, in which the mice would experience sudden narcoleptic attacks with a characteristic collapse of the head and neck and buckling of the limbs. EEG studies ruled out the possibility of seizure disorders and confirmed a disruption of REM sleep.
There has been a flurry of activity since the Hcrt-narcolepsy link was made in 1999. The search for genetic alterations in human narcolepsy has demonstrated that
Hcrt and
Hcrtr mutations are rare. This is not surprising as most human narcolepsy is not familial and is discordant in identical twins. In 2000, Mignot’s group reported one
Hcrt mutation in a case of early onset narcolepsy out of 74 narcoleptic patients studied.
6 What they did find, however, was a global deficiency in Hcrt. In situ hybridization studies revealed an absence of
Hcrt mRNA in the hypothalami of narcoleptic patients, and radioimmunoassays showed low levels of Hcrt in the CNS. This was consistent with a previous report from Mignot’s group that 7 out of 9 narcoleptic patients studied had low CSF Hcrt.
7 The likely cause of narcolepsy in humans was found to be a degeneration of Hcrt neurons. An immunohistochemical study of 16 human hypothalamus samples, four narcoleptics, and 12 neurologically healthy subjects, revealed an 85% to 95% reduction in the number of Hcrt neurons in narcoleptic hypothalami.
8 The neuronal loss was limited to Hcrt neurons as melanin-concentrating hormone (MCH) neurons, which are intermixed with Hcrt cells, were unaffected. This raised the possibility of narcolepsy being a neurodegenerative disease such as Alzheimer’s disease or Parkinson’s disease. The association between narcolepsy and HLA-DQB1*0602 made autoimmune disease a possible etiology but to this day, little evidence of immune-mediated neuronal degeneration has been found.
9 However, brain samples are often examined late in the disease process, possibly past the point of detecting any sign of autoimmune disease. Despite strong evidence from animal and human studies that hypocretin plays a role in the development of narcoleptic symptoms, the exact pathophysiological link between hypocretin and narcolepsy in humans remains unclear. Animal data clearly demonstrate that
Hcrt knockout mice develop narcolepsy-like symptoms, and the canine model of narcolepsy involves mutated
Hcrt receptors. However, the disease process is clearly different in humans and one cannot make inferences based on the animal data. In addition, a recent study showed that 11% of narcoleptics with cataplexy have normal CSF Hcrt levels.
10 What the authors do know is that most humans with narcolepsy have a reduction in Hcrt-producing neurons in the hypothalamus, but the authors do not know whether this is causative of the disease. Studies performed in recent years have demonstrated that there is much more to Hcrt function than sleep regulation (
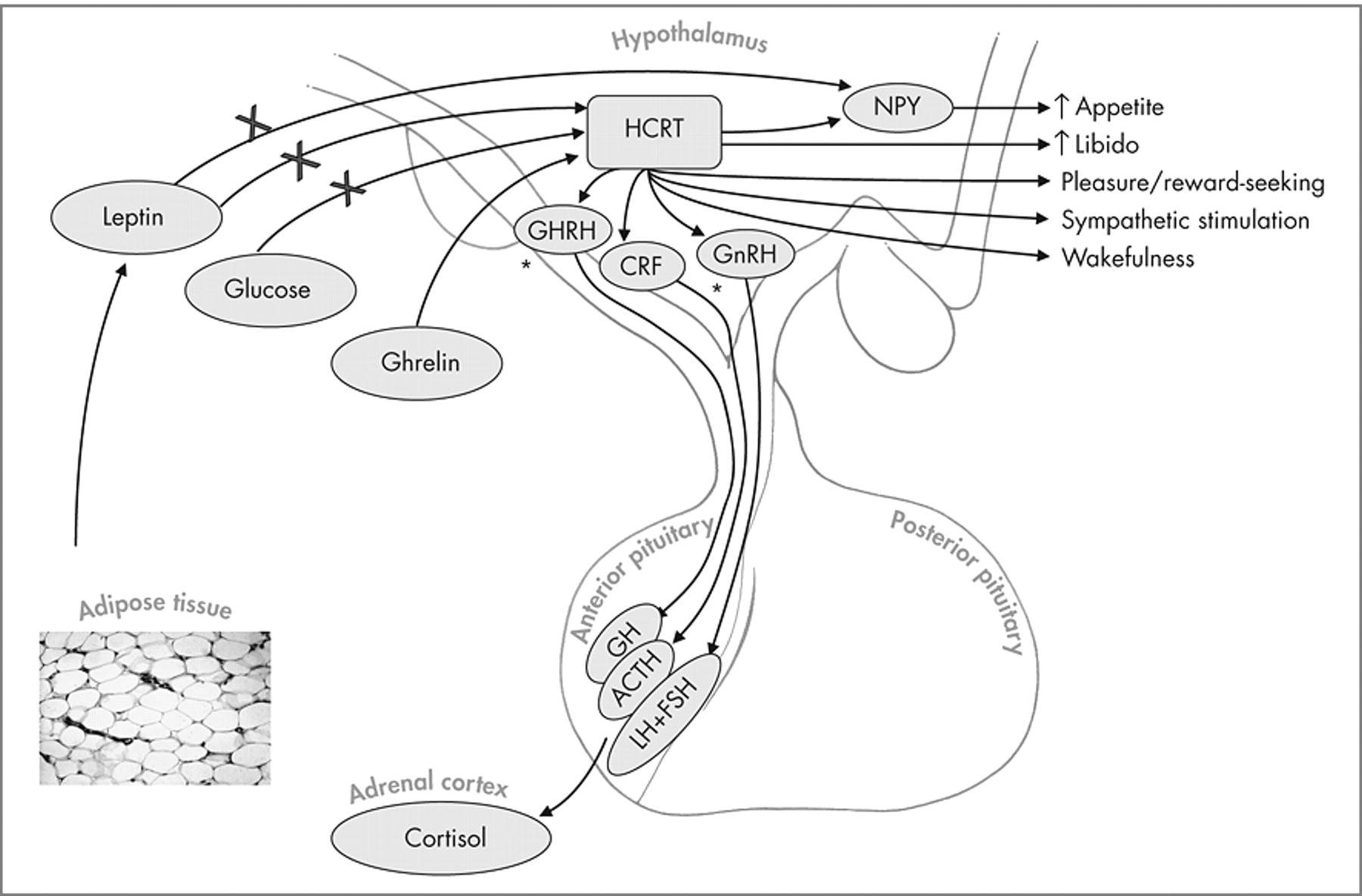
Figure 1 ) and that the Hcrt peptide system is altered in disease processes other than narcolepsy. Hcrt has a number of autonomic and endocrine functions and has been shown to influence cardiovascular function, reproductive and stress hormone secretion and appetite and energy balance.
11 In addition to narcolepsy, decreased levels of Hcrt have been found in patients with Guillain-Barré syndrome (GBS), acute lymphocytic leukemia (ALL), intracranial tumors, craniocerebral trauma, and CNS infections.
12Autonomic Functions of Hypocretin
When exogenous Hcrt was administered to reverse the symptoms of narcolepsy, the animals experienced an increase in blood pressure and heart rate.
13,
14 This is not entirely surprising as regions of the hypothalamus and brainstem that are involved in cardiovascular regulation contain Hcrt neurons and express Hcrt receptors.
2,
15 These effects are caused by activation of the sympathetic branch of the autonomic nervous system (ANS)
13,
14 but it is also possible that Hcrt can influence the dorsal motor nucleus of the vagus nerve, which is a major parasympathetic center.
16 The ability of Hcrt to stimulate sympathetic activity is fairly well established, however, some groups report depressor effects especially when Hcrt is directly injected into other regions of the brain. de Oliveira et al.
17 report that Hcrt-1 in the nucleus tractus solitarus (NTS) increases vagal activity to the heart and inhibits sympathetic activity. The same group reported similar findings with Hcrt-1 in the nucleus ambiguus.
18 These contradictory findings can be explained by the inherent limitation of in situ experiments with neurotransmitters. Site-specific delivery is not anatomically localized or functionally specific and does not represent true physiological conditions. In vivo experiments seem to indicate that Hcrt has primarily a sympathostimulatory effect. Knockout mice deficient in Hcrt have a significantly lower mean arterial blood pressure (MAP) than normal mice. The difference in MAP in wild-type and mutant mice can be cancelled with α-adrenergic blockade and ganglionic blockade. These treatments lowered the MAP of wild-type mice significantly more than that of mutant mice resulting in MAPs that were statistically identical. Conversely, treatment with angiotensin-converting enzyme (ACE) inhibition or vasopressin antagonism affected the MAP of both wild-type and mutant mice in a similar manner. This suggests that the cardiovascular status of the knockout mice is due to decreased sympathetic vasoconstrictor tone.
19 In human narcoleptics, subnormal heart rate, blood pressure, and forearm blood flow responses to muscle contraction are observed as well as subnormal heart rate response to the Valsalva maneuver.
20Endocrine Functions of Hypocretin
Experiments involving direct injection of Hcrt into animal brains, including the hypothalamus, have shed light into its role in modulating neuroendocrine pathways. The presence of Hcrt-positive axons in regions of the brain important in anterior pituitary function and the presence of Hcrtr-1 and Hcrtr-2 in the pituitary glands of rats suggest that Hcrt could play an important role in the hypothalamo-pituitary-axis.
21 To date, Hcrt has been found to modulate the release of the hypothalamic hormones, gonadotropin releasing hormone (GnRH), and corticotropin releasing factor (CRF). GnRH-producing neurons express Hcrtr-1 in rat hypothalamus and 75% to 85% of these neurons come in contact with Hcrt fibers.
22 Hcrt-1 has been shown to stimulate GnRH release from rat hypothalamic explants in vitro and modulate LH release in vivo.
23 It remains doubtful, however, that these interactions are physiologically relevant as Hcrt knockout mice are fertile,
5 implying that their LH and FSH levels are normal, despite the fact that they lack Hcrt. Other studies have linked Hcrt to regulation of prolactin (PRL) and growth hormone (GH) release, but these interactions also have yet to be shown to be physiologically relevant. One neuroendocrine interaction that may be important is Hcrt’s effect on stress hormone secretion. Central administration of Hcrt in rats results in increased plasma adrenocorticotrophic hormone (ACTH) and corticotropin-releasing factor (CRF); this increase does not occur if the animal is pretreated with a CRF antagonist.
24,
25 These animals become obese and are at an increased risk of developing diabetes, as is the case in humans with excess corticosteroid.
Appetite Regulation
The location of Hcrt-positive neurons, namely the lateral hypothalamus, is also known as the “hunger center” of the brain. Lesions in this region decrease eating behavior while stimulation increases eating. Hcrt itself acts as a weak-to-moderate appetite stimulator, but it also interacts with other neuropeptides to modulate feeding behavior. The most convincing evidence of Hcrt’s role in feeding behavior comes from mouse studies in Sakurai’s laboratory. Their group generated transgenic mice in which Hcrt-containing neurons are ablated by expression of a disease gene product, ataxin-3, that is only expressed in Hcrt-containing cells. Over time, these mice become obese, despite eating less, much as human narcoleptics have been described to do.
26Neuropeptide Y (Npy) is widely recognized as an extremely potent stimulator of feeding behavior and its blockade by antibody or antisense RNAs(1) disrupts this behavior. Many groups have shown that the effect of Hcrt on feeding is, at least in part, mediated by Npy. Hcrt-positive axons connect to Npy neurons in the arcuate nucleus and the Npy neurons express Hcrt receptors.
27 Lopez et al.
28 demonstrated that administration of Hcrt transiently increased Npy expression in the arcuate nucleus of rat hypothalamus. Pretreatment with an Npy antagonist abrogated the appetite-stimulatory effect of Hcrt suggesting the Npy pathway is downstream from Hcrt. Also upstream from Npy is a protein hormone called leptin. Leptin is primarily expressed by adipocytes which allows the body to produce a measure of nutritional status. Leptin receptors are primarily expressed in the hypothalamus. Increased leptin expression decreases synthesis of Npy and, thus, inhibits feeding. Hcrt neurons also express leptin receptors and appear to be regulated by leptin. Human narcoleptics matched for age, sex, body mass index, waist/hip ratio, and fat mass have been shown to have plasma leptin levels about half of those found in control subjects, possibly due to sleep/wake disturbances.
29 Leptin also regulates energy expenditure as measured by oxygen consumption and body temperature. This is consistent with what is observed in human narcoleptics who eat less but still become obese, as well as the Sakurai mice that lose the Hcrt neurons that express the leptin receptor.
26 Decreased Hcrt levels decrease appetite via down-regulation of Npy while increased weight gain may be a result of decreased energy expenditure and metabolism from low circulating plasma leptin. Sakurai’s group has also characterized other metabolic cues that regulate Hcrt neuron activity. Glucose, leptin, and monoamines inhibit Hcrt neurons while ghrelin, a feeding-stimulatory peptide that rises during fasting, stimulates them. The discovery and characterization of the Hcrt peptide system demonstrates the intricate relationship between wakefulness and feeding. Its purpose may be to promote wakefulness when facing an energy shortage.
Prader-Willi Syndrome
There are a number of neurological disorders that exhibit both sleep and appetite disturbances in which Hcrt function is potentially an important factor. Prader-Willi syndrome is a genetic disease characterized by hypotonia, developmental delay, failure to thrive, increased appetite, and childhood obesity. Sleep disturbances are also common in Prader-Willi syndrome cases. Excessive daytime sleepiness is a common symptom of Prader-Willi syndrome
33 and sleep disordered breathing, sleep-onset REM periods, and cataplexy have also been reported
34,
35 suggesting possible common pathways between narcolepsy and Prader-Willi syndrome. Though there have been no large studies examining the Hcrt system in Prader-Willi syndrome, a number of smaller studies exist which make the case that disruption of the Hcrt system can explain some of the symptoms observed in this disorder. A recent study by Nevsimalova et al.
36 looked at CSF Hcrt-1 levels in four Prader-Willi syndrome patients. All four patients were extremely obese, two of the four had excessive daytime sleepiness and none had cataplexy. The two cases with excessive daytime sleepiness were found to have the lowest levels of Hcrt (approximately half of normal) while the other two had intermediate or normal levels compared to healthy comparison subjects. Arii et al.
37 reported a 2-week-old baby with severe Prader-Willi syndrome who had intermediate levels of Hcrt. Traditionally, the sleep disturbances seen in Prader-Willi syndrome were thought to be due to weight, but these studies indicate that Hcrt could also play an important role. Similarly, Hcrt could explain the sleepiness experienced by obese women who do not have obstructive sleep apnea.
38 The mechanism for disruption of the Hcrt system appears to be different in Prader-Willi syndrome patients versus narcoleptics however. While narcoleptics appear to have slow degeneration of Hcrt neurons over time, possibly from an auto-immune reaction, Prader-Willi syndrome patients have no change in the number of Hcrt neurons, as recently reported by Fronczek et al.
39 The differing mechanisms for the disruption of the Hcrt system could also explain why the narcolepsy-like symptoms in Prader-Willi syndrome patients differ slightly from those seen in narcoleptics. For example, while both groups have a tendency toward obesity, narcoleptics tend to eat less than healthy subjects while Prader-Willi syndrome patients tend to eat voraciously.