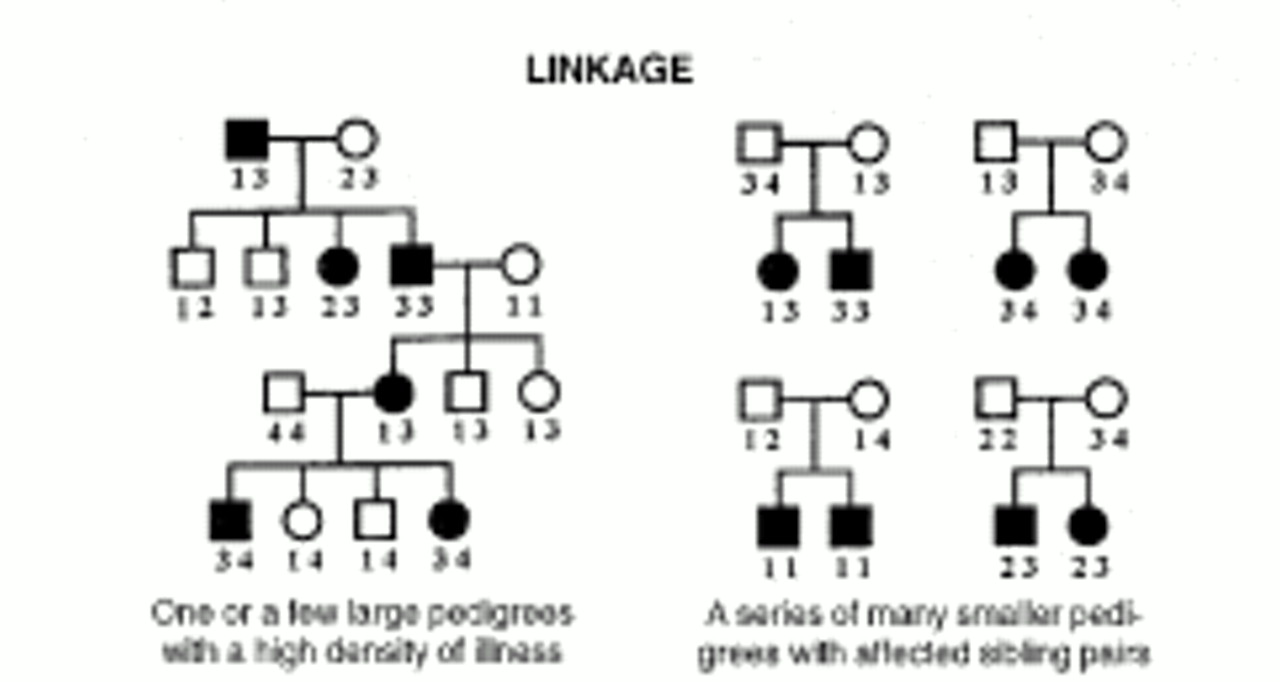
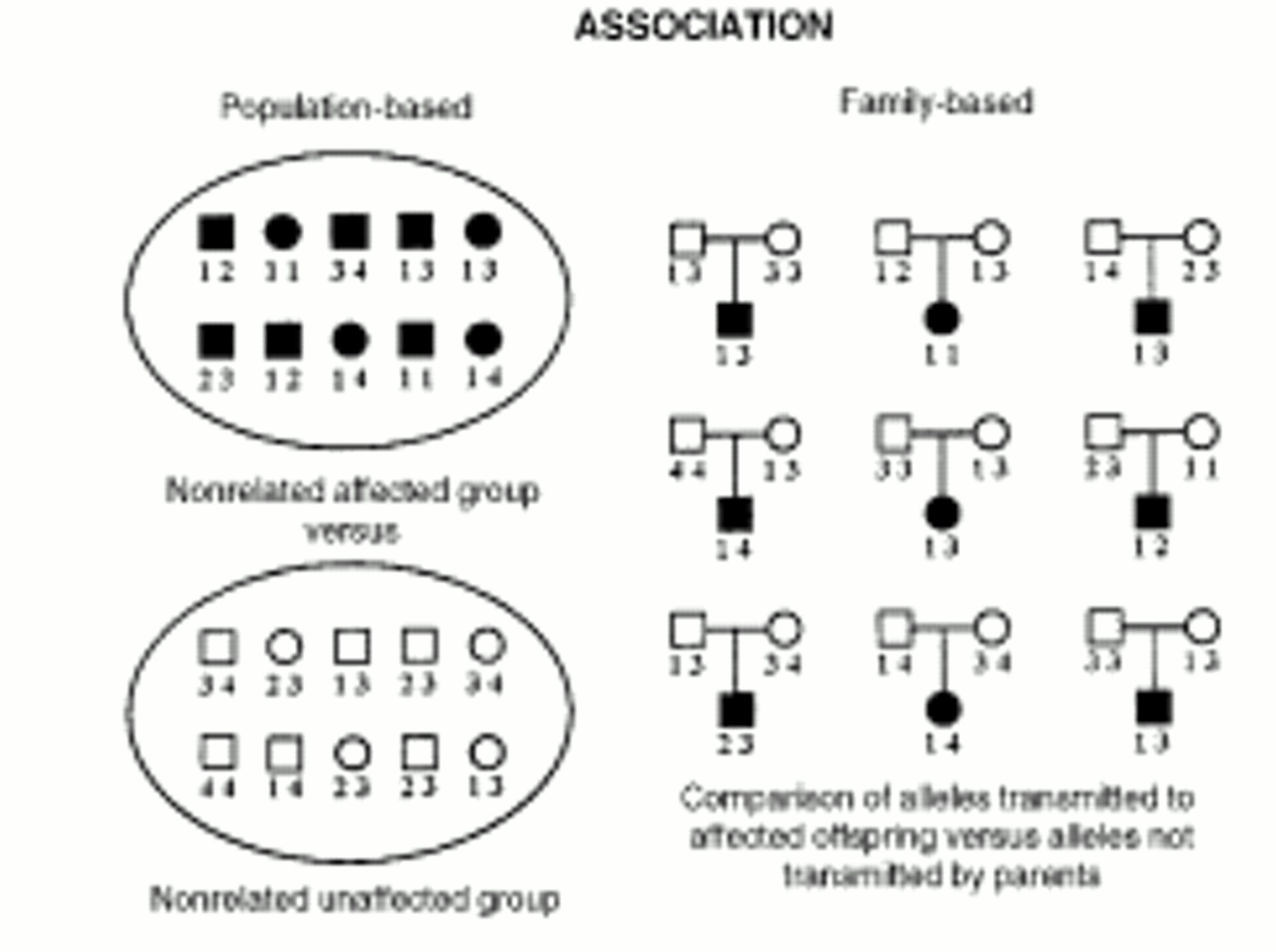
Complex genetic illnesses are familial disorders that are not attributable to a single dominant or recessive gene but rather to several genes, which contribute to the illness in a multiplicative or additive fashion (polygenic inheritance). The genes involved in complex genetic diseases are harder to detect because of the number involved and the generally small effect of each component gene. Most common diseases, including schizophrenia and bipolar disorder, fall into this category.
A parametric (inheritance model-dependent) type of linkage analysis is the logarithm of the odds ratio (LOD) score analysis, which estimates the likelihood of the observed distribution of markers and illness in a family under a predetermined model of inheritance. For simple Mendelian diseases, a LOD score statistic of 3.0 or higher in a genome-wide scan is considered a statistically significant linkage (p<0.05). For psychiatric diseases, however, a model of inheritance must be assumed, since a correct single-gene model of inheritance has not been demonstrated. Also, disease-causing alleles may have a high frequency in the population yet a low penetrance. This can interfere with traditional linkage analysis, since the same gene can be introduced by several ancestors or persons marrying into the family. These uncertainties undermine parametric analyses.
Allele-sharing methods, which are nonparametric (inheritance model-independent), tend to be a more robust type of linkage analysis. The affected sibling pair method measures the frequency that both siblings inherit the same genetic marker allele (or variant) from each parent (referred to as identical-by-descent, or IBD). Presence of a disease-causing gene is revealed when the IBD allele-sharing between affected siblings exceeds the expected 50%.
As the linkage evidence solidifies for certain chromosome areas that contain susceptibility genes for psychiatric illness, efforts will shift toward narrowing down the currently rather large linkage regions, which in some instances can be done through association. All association studies are properly based on linkage disequilibrium. This means that an illness gene mutation initially occurred close to specific alleles of nearby polymorphic markers. As generations (and meioses) proceed over time, the illness gene and marker allele remain statistically associated because their proximity markedly reduces the number of recombinations (crossing over) that occurs between them. Statistically, association studies are often more powerful than linkage analyses in that a valid association may be detected in a sample in which linkage is not detectable, even when the gene is playing only a modest role in illness susceptibility. This is especially likely when the marker assayed is the susceptibility gene.
Most association studies in the past were population-based: allele frequencies of a nonrelated affected group were compared against those of a nonrelated unaffected group. However, with the discovery that racial and ethnic groups display different allele frequencies at many polymorphic markers, it became apparent that stratification often led to artifactual allele frequency differences between the affected and unaffected groups. Current association studies are family based, which avoids this artifact.