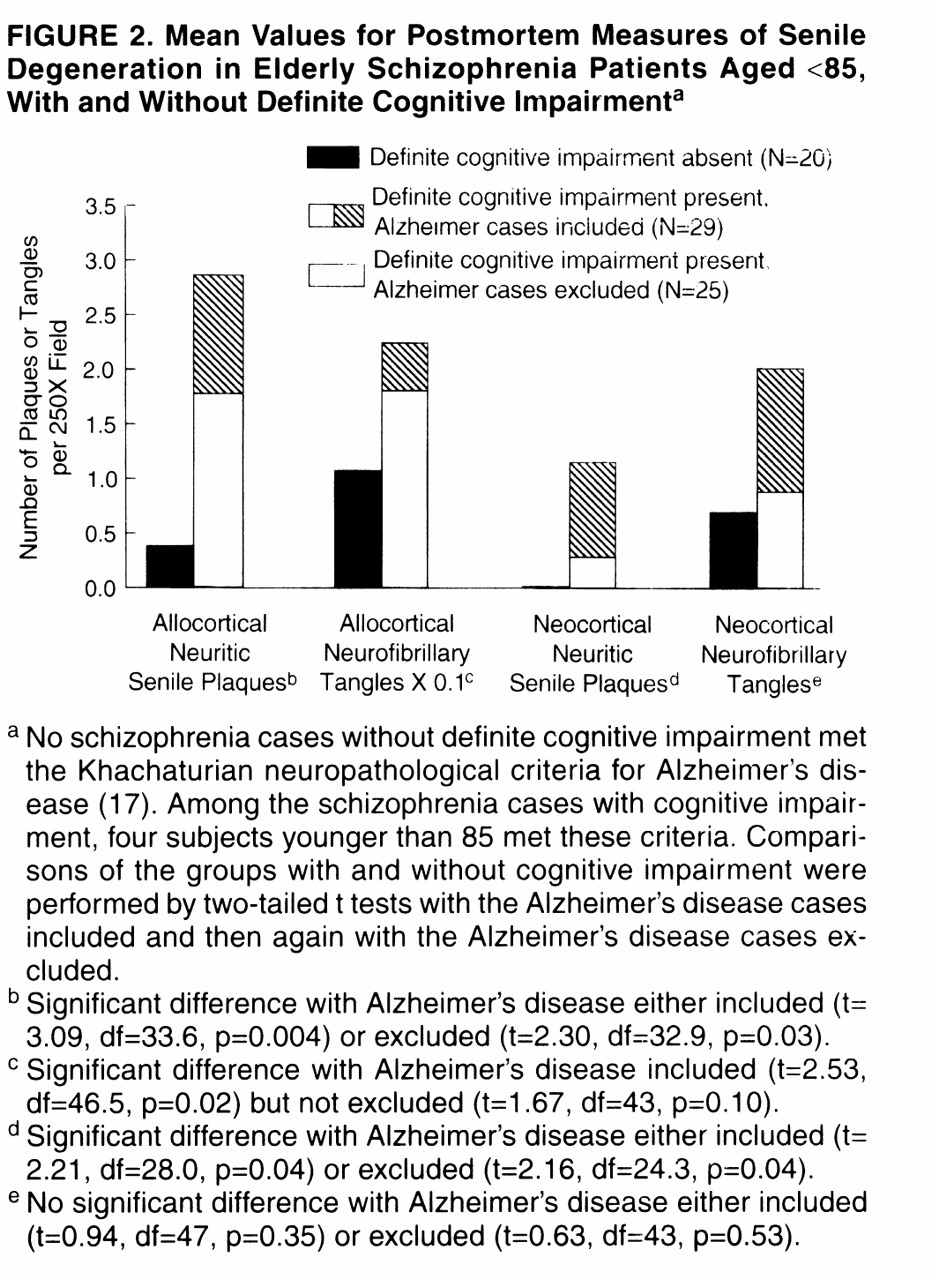
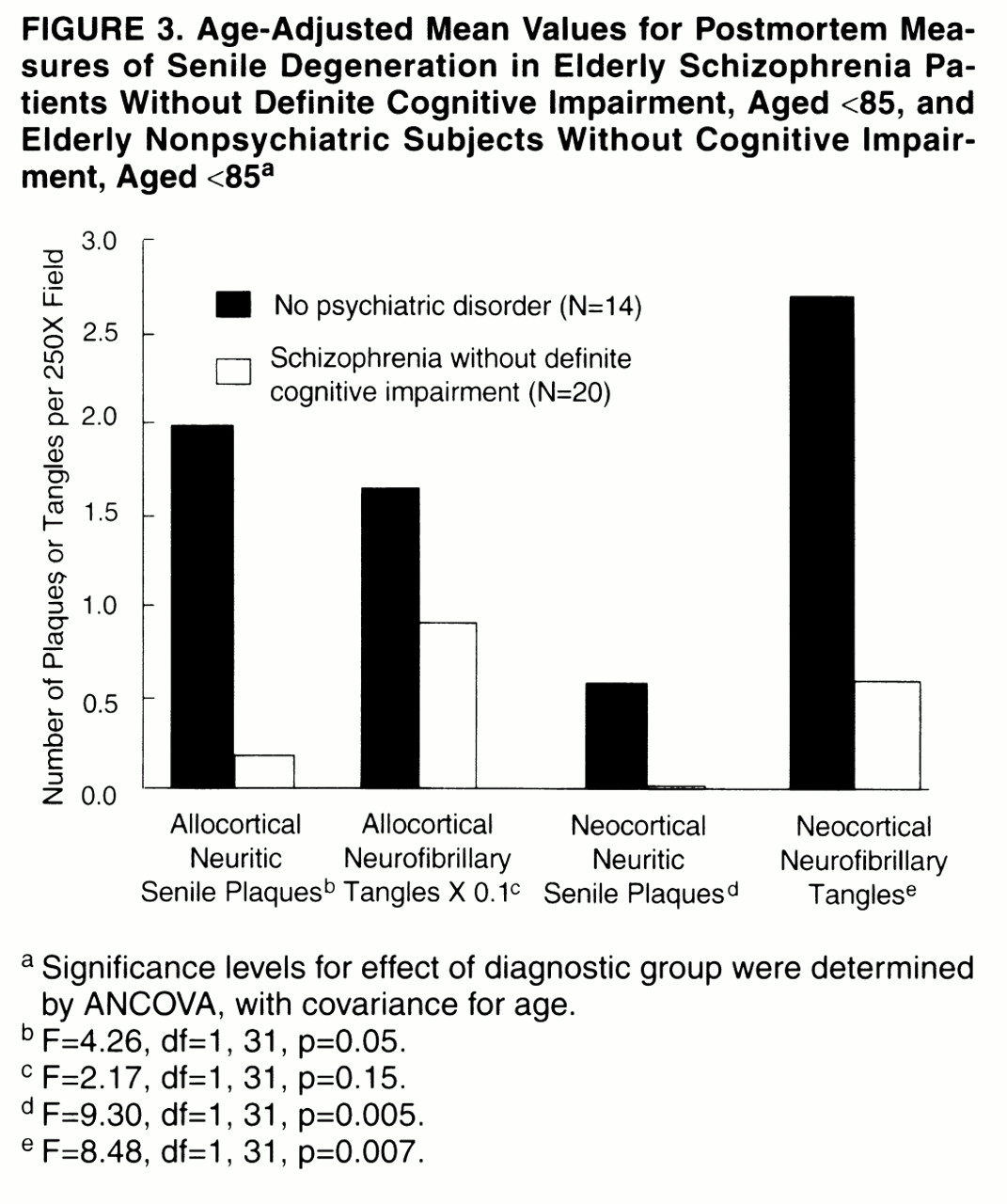
Our study yielded four main findings.
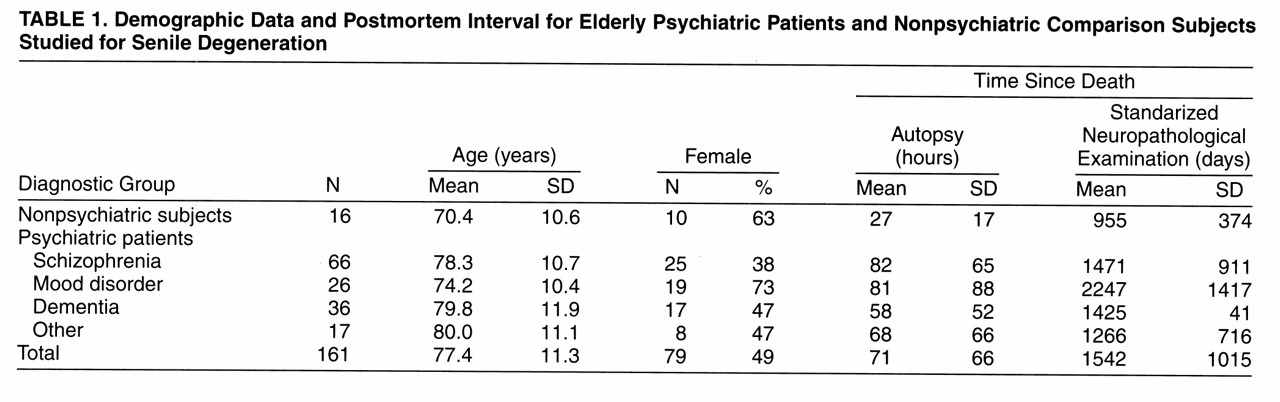
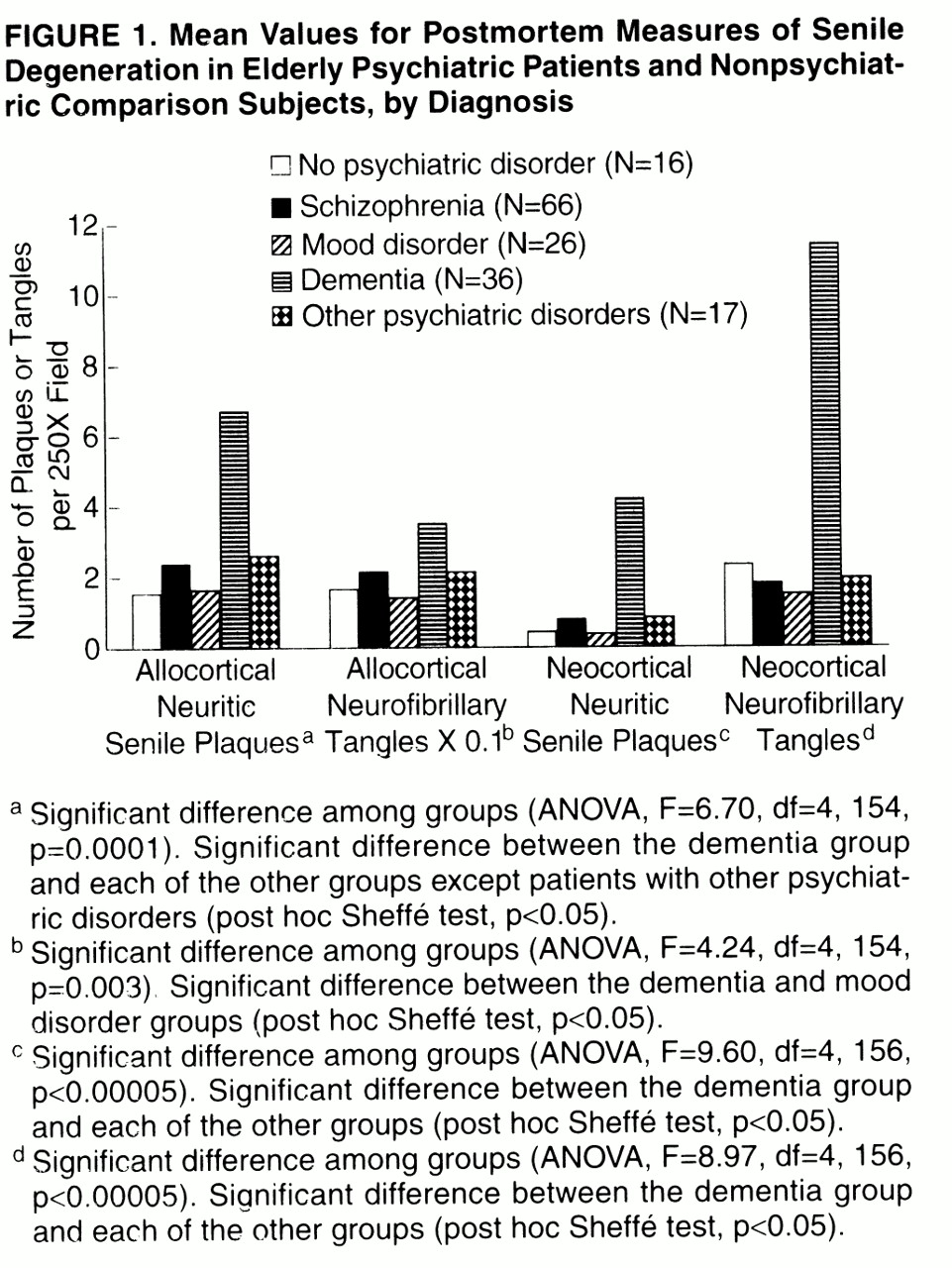
A parsimonious interpretation of these findings is that schizophrenia creates a vulnerable state in which even mild senile degeneration, particularly in the form of neocortical neuritic senile plaques, is sufficient but not necessary to cause definite cognitive impairment. In contrast, mild levels of senile degenerative change appear to be better tolerated in subjects without preexisting psychiatric disease. For example, four of 16 normal subjects had at least four neuritic senile plaques per field in at least one neocortical region. However, while the nonpsychiatric subjects clearly lacked the behavioral manifestations of cognitive impairment that were present in the subjects with schizophrenia, neuropsychological testing or detailed clinical evaluation may elicit subtle cognitive differences between elderly subjects with and without mild Alzheimer-type changes
(20, 24, 25). Individuals who can tolerate such cognitive loss without behavioral manifestations must posses either compensatory abilities or a surfeit of cognitive ability. We suggest that these are lacking in chronically institutionalized subjects with schizophrenia.
Our findings are similar to those of El-Mallakh et al.
(26), who found more frontal and hippocampal senile plaques in 10 intellectually impaired schizophrenia subjects (mean age=84 years) than in seven intellectually intact schizophrenia subjects (mean age=62). In striking similarity to our study, the mean plaque counts for the intellectually intact schizophrenia subjects were extremely low, approximately one-tenth of the mean value for seven normal subjects (mean age=73).
Two recent studies of elderly, chronic inpatients had findings somewhat different from ours. One study
(10) showed levels similar to ours for neocortical neuritic senile plaques in nonpsychiatric subjects and cognitively impaired schizophrenia subjects but a higher value for cognitively unimpaired schizophrenia subjects, close to the value for the nonpsychiatric subjects. All ages were similar, and we cannot attribute this difference to staining or counting procedures, since the values were similar for the first two groups, nor to overdiagnosis of definite cognitive impairment on our part, since Purohit et al.
(10) classified 87 of 100 schizophrenia subjects as cognitively impaired, while we classified 68% as having definite cognitive impairment. Purohit et al. counted neuritic senile plaques in the orbitofrontal cortex, while we did not. If their higher value was attributable to neuritic senile plaques in this region, it is possible that these are better tolerated than neuritic senile plaques in the temporal neocortex. In another study
(27) there was no correlation between any neuropathological measure and any cognitive measure in 23 elderly schizophrenia subjects. Since that study did not include counts of
neuritic plaques, it is difficult to compare with ours.
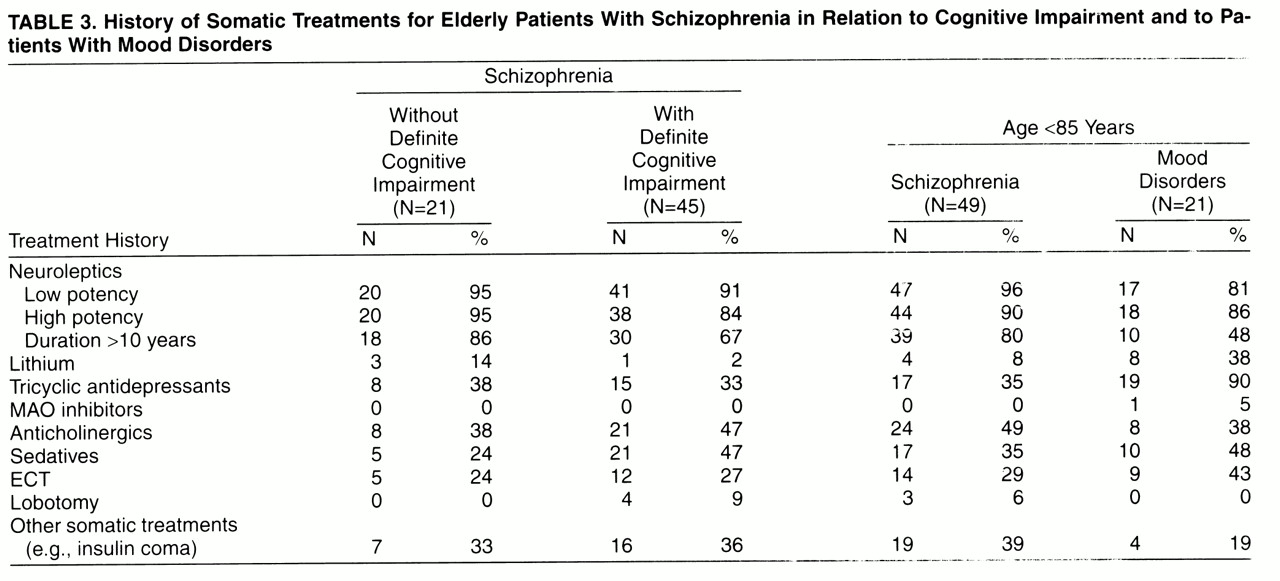
Enhanced sensitivity of cognition to neuritic senile plaques was not apparent in our group of chronically institutionalized patients with mood disorder, although this group was similar to the schizophrenia group in terms of age, presence of psychosis, institutionalization, and somatic treatments. The subjects in the two groups died during the same period and in the same institutions; hence, one should expect similar reporting of their symptoms in the medical records. Thus, from the available data, we cannot attribute the high rate of cognitive impairment in schizophrenia to institutionalization, somatic treatments, or reporting biases of clinicians.
“Cognitive Reserve” in Schizophrenia
Our findings suggest that schizophrenia and senile degeneration are synergistic in producing cognitive decline. This interpretation is consistent with theories of “cognitive reserve” protecting against dementia, first proposed by Katzman et al.
(30). These investigators found that the brains of nondemented elderly individuals with large numbers of neuritic senile plaques but no dementia were heavier and contained more neurons than those of elderly individuals with dementia. Evidence that higher educational and occupational levels protect against the dementing effects of neuritic senile plaques and neurofibrillary tangles has been interpreted as further support for the cognitive reserve hypothesis
(14, 15). The observation that neocortical synaptic density provided the best neuroanatomic correlate for cognitive function in Alzheimer’s disease
(31, 32) has led to the hypothesis that the protective effect of education resides in increased synaptic density
(14). Under this model, we postulate that senile impairment of cognition in schizophrenia and nonschizophrenia subjects alike results from a continuous, age-related process that is correlated with the appearance of neuritic senile plaques. The threshold at which this process produces clinical evidence of cognitive change is hypothetically lower in individuals with schizophrenia than in others.
The underlying vulnerability in schizophrenia may be developmental; several investigators have reported low cognitive capacity in young subjects
(3, 33, 34). Vulnerability could also be associated with some progressive process, yet unidentified, that is specifically related to schizophrenia. Theoretically, such a process could produce cognitive impairment by itself when sufficiently advanced, or at an earlier stage if neocortical neuritic senile plaques are also present.
Processes contributing to diminished cognitive reserve in schizophrenia have yet to be identified. In contrast to Alzheimer’s disease, where loss of synaptic density
(31, 32) and cholinergic markers
(35) are prominent features and correlated with cognitive impairment, diminished synaptic density has not been reported in schizophrenia, and cholinergic markers are normal
(26, 35). However, reported abnormalities in cortical synaptic proteins
(36–42), microtubule-associated proteins
(43–45), neuronal size
(46–48), and neuropil volume
(49) suggest the possibility of impaired synaptic function or remodeling.
Dopaminergic deficits are associated with dementia in a variety of conditions
(50–55). Although the positive symptoms of schizophrenia have generally been related to subcortical dopaminergic hyperactivity, many authors have suggested that in schizophrenia, some aspects of dopaminergic activity are suppressed
(56–59), which could contribute to cognitive impairment
(3). Low
(60) and normal
(61) prefrontal immunoreactivity for tyrosine hydroxylase have both been reported. If dopaminergic reserves are even focally diminished in schizophrenia, further decline with age (normally beginning in adolescence
[62] and progressing through senescence
[63]) could impair cognition or increase vulnerability to the effects of other aging processes.
Neuroleptic drugs could, of course, play a part. They are antagonists at a variety of receptors for dopamine, acetylcholine, serotonin, norepinephrine, and epinephrine. The authors of a report of low tyrosine hydroxylase activity in the deep layers of the anterior cingulate cortex noted the absence of this abnormality in two neuroleptic-free patients
(61), raising the possibility that neuroleptic drugs influence the distribution of cortical dopamine terminals. However, the relatively low incidence of cognitive impairment in the neuroleptic-treated mood disorder patients in our study indicates that neuroleptics alone do not account for the greater sensitivity to neuritic senile plaques and neurofibrillary tangles that we find in schizophrenia.