Genetic transmission in major depression is well documented, although common disorders, such as depression, are genetically heterogeneous (
1). Imposing a genetically mediated physiological measure on those at risk of depression can reduce heterogeneity. Such a characteristic can simplify phenotypes, improve detection of heritability, and increase detection of at-risk individuals. Measures of polysomnographic sleep can serve these functions, according to extensive evidence of a genetic influence on sleep (
2–
6) and on well-described sleep abnormalities in unipolar depression.
A reliable constellation of polysomnographic abnormalities has been observed in depressed patients, including disrupted sleep, decreased slow wave sleep, and REM sleep dysregulation (
7,
8). The specificity of a single abnormality has been challenged (
8), yet the constellation of abnormalities has been relatively specific as well as robust (
9,
10). For example, in preliminary work we found that affected, but currently asymptomatic, relatives of depressed probands with short REM latency had other polysomnographic alterations consistent with those found in depressed patients, including decreased slow wave sleep and increased REM density (
11). Of critical importance, this pattern was also found among a small sample of relatives with no personal history of depression. Polysomnographic abnormalities in unaffected relatives suggest that a sleep signature may identify unaffected individuals at risk of depression.
We now have a substantial cohort of families identified by depressed probands with short REM latency, depressed probands with normal REM latency, and normal comparison probands. Our hypotheses were that 1) polysomnographic abnormalities, specifically short REM latency, will occur with increased frequency within families of affected probands, and 2) short REM latency will be associated with increased risk of depression. The rationale for using REM latency as the sole physiologic characteristic followed from earlier work confirming its utility. Short REM latency is a reliable correlate of unipolar depression (
12–
14), stable during an episode (
15) and stable during remission (
16–
19). We also found that it was concordant in first-degree relatives with the same type of depression (
20,
21). REM latency, however, is only one of several potentially informative sleep variables. In the present report, we evaluated the concordance of polysomnographic abnormalities in a large group of families of unipolar probands and controlled for the familial nature of sleep by including normal comparison families. Accordingly, this report extends our previous work by including a matched, normal comparison group and fuller representation from families.
We tested for sleep dysregulation and for the cosegregation of sleep dysregulation and unipolar depression by assigning depressed probands to “short” and “normal” groups on the basis of REM latency. In the normal comparison group, psychiatric history was controlled and REM latency was free to vary. We therefore had comparison groups for short REM latency and for psychiatric history.
We planned several steps to test for cosegregation of sleep abnormalities and depression. First, we tested our hypothesis of greater frequency of polysomnographic abnormalities among relatives of depressed probands with short REM latency than among relatives of depressed probands with normal REM latency and the normal comparison group. Second, we tested the hypothesis that short REM latency conferred increased risk of depression within depressed families. Third, we determined the sleep profile of relatives on the basis of proband REM latency and the additive value of other EEG sleep measures in predicting lifetime depression. Fourth, we determined whether short REM latency occurred within unaffected relatives in depressed families and could index increased risk of depression. These hypotheses are addressed in this report. We have also monitored the clinical course of unaffected and affected relatives to determine whether polysomnographic abnormalities prospectively predicted onset of depression and morbidity in the clinical course. These prospective studies are ongoing.
METHOD
Subjects
Unipolar depressed and normal comparison probands. Data on 17.1%–23.8% of participants have been reported previously. In 1988 (
21), we described the relationship between short REM latency in 14 probands and rate of major depression in 43 parents and siblings. In 1989, we commented on the secular trend in depression, using data from 23 probands and 60 relatives (
22). We also evaluated polysomnographic data of 54 relatives of depressed probands with short (N=10) and normal (N=13) REM latency (
11).
Depressed probands were recruited from research protocols on the basis of the following: 1) unipolar depression (Research Diagnostic Criteria [RDC] and DSM-III-R); 2) two living biological parents and living, fully consanguine siblings; 3) no major medical illnesses or primary sleep disorders, and 4) no bipolar disorder or schizophrenia in first-degree relatives. Normal comparison probands were recruited from advertisements and research protocols. Identical criteria were used except that probands reported no personal or family history of psychiatric disorders in first-degree relatives. Normal probands were group-matched to depressed probands with short REM latency by proband age and sex and by age and size of families. Written informed consent was obtained after complete description of the study to all subjects.
Probands were interviewed with the Schedule for Affective Disorders and Schizophrenia—Lifetime Version (
23). Symptom severity (Hamilton Rating Scale for Depression [24]), episode, treatment, medical history, and personal and family history of sleep disorders were also ascertained. Family history of psychiatric disorders was obtained according to Family History Research Diagnostic Criteria (FH-RDC) (
25).
First-degree relatives. Relatives were consanguine parents and siblings, as ascertained by history. The design to recruit parents and adult siblings allowed full sampling of the families, which controlled for age of risk, and allowed psychiatric history to vary in families of depressed probands.
After complete description of the study to relatives, written informed consent was obtained. Relatives were evaluated following procedures identical to those used with probands. Consensus diagnoses were derived for noninterviewed relatives by using the multiple informant procedure, based on FH-RDC.
Relatives were eligible for sleep laboratory evaluation if they followed a diurnal sleep/wake schedule and were medically healthy or if neither the illness nor the treatment altered polysomnography results (e.g., thyroid replacement hormones; antibiotics for acne). Twenty-one relatives were interviewed but were medically inappropriate or unwilling to be studied in the laboratory.
Most relatives (95.6%) had no acute psychiatric illness at evaluation, regardless of lifetime history; 90.5% of relatives had Hamilton depression scale scores of 7 or less. Because mood-related symptoms affect several polysomnographic measures, however, we compared relatives who had Hamilton depression scores of 7 or more (N=20, mean Hamilton depression score=12.1, SD=4.2) with relatives who were well and had Hamilton depression scores of 7 or less (N=196). There were no sleep or clinical differences, and these data were included in analyses.
Procedure
Subjects completed a sleep diary for 14 days before sleep study and reported sleep onset time, bedtime, rise time, naps, medications, exercise, and meals. Subjects were instructed to 1) maintain consistent bedtimes and rise times, 2) refrain from napping, and 3) discontinue medication. Medications included alcohol, psychotropic drugs, and over-the-counter products except routine vitamins and acetaminophen. Participants had not been treated with monoamine oxidase inhibitors or selective serotonin reuptake inhibitors. Five days before laboratory assessment, subjects were asked to discontinue caffeine products. Subjects were asked not to exercise before recordings. Probands were studied in air-conditioned, sound-attenuated bedrooms in the sleep laboratory for a minimum of 2 nights. A few depressed probands (15%) were studied as inpatients, and prelaboratory monitoring could not be followed. These patients observed hospital schedules for a minimum of 7 days. First-degree relatives were studied for 3 consecutive nights. Subjects 55 years of age and older and subjects with clinical evidence of sleep disorders were screened the first night with the appropriate montage and were excluded if a sleep disorder was found.
Polysomnographic data included the EEG, electro-oculogram, and electromyogram. We used Grass Model 78 polygraphs with time constant of 0.3 msec and sensitivity of 5 for the EEG, calibration of 50 μV/10 mm, and paper speed of 10 mm/second. Data were scored in 30-second epochs (
26). Sleep onset was defined as the first half-minute of 10 consecutive minutes of any stage sleep interrupted by no more than 2 minutes of awake or movement time. REM latency was calculated as time from sleep onset to the first 30-second REM epoch. REM density was scored on a 0–8 scale per 60-second epoch. Data from nights 2 and 3 were used for analyses if there were no technical difficulties. We compared sleep data between subjects for whom nights 2 and 3 were used and subjects for whom technical difficulties required use of alternative nights. No differences on any measures were found.
Data Analyses
Data from four depressed families (two with short and two with normal REM latency) were excluded from analyses. In two cases, prospective follow-up revealed use of psychoactive substances during the 14-day period before sleep study; in the third case, parents had primary sleep disorders, as determined through laboratory evaluation. In the fourth case, the proband revealed a primary diagnosis of alcoholism in follow-up.
We used our matched-group design to identify an appropriate threshold.
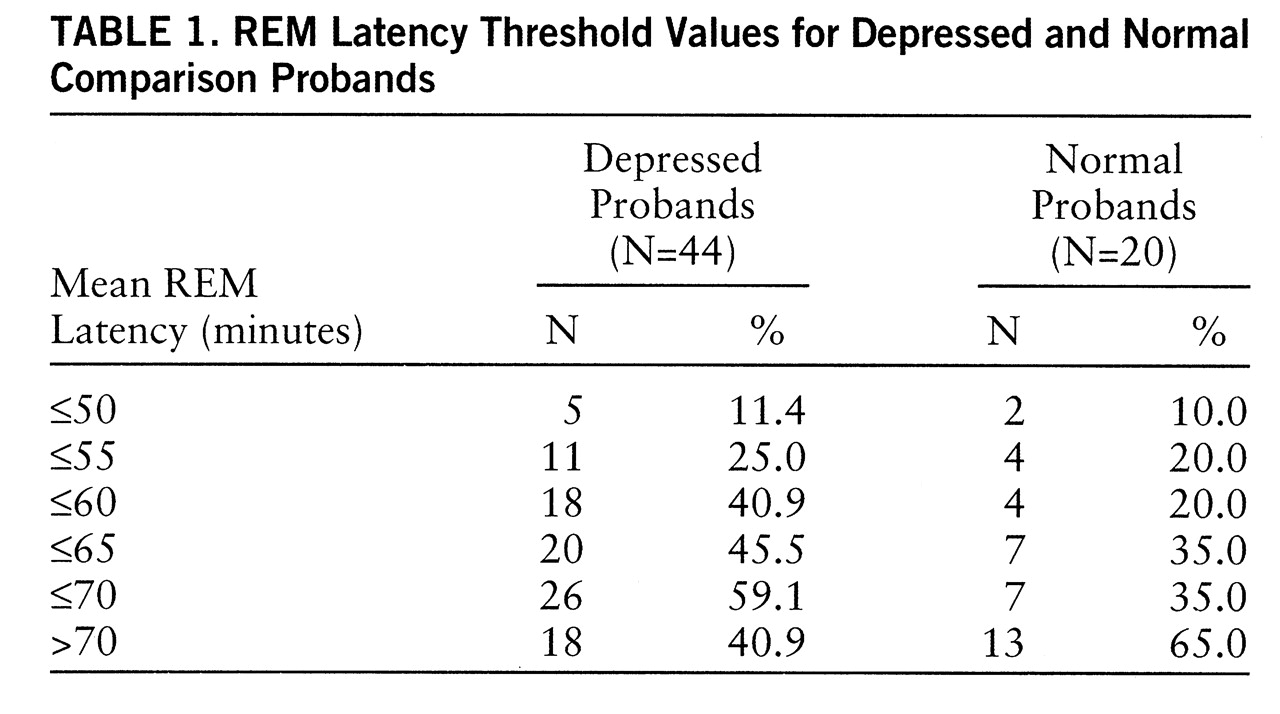
Table 1 presents proportions of depressed (N=44) and normal (N=20) probands with mean REM latency at a series of threshold values. The frequency of REM latency ≤55.0 minutes was low (20%–25%) for both proband types. Within the normal group, sensitivity did not differ between 55 and 60 minutes, whereas the number of depressed probands identified by REM latency ≤60 minutes doubled. Using the criterion value of 60 minutes, we found that 18 depressed probands had short REM latency and 26 had normal REM latency. Among the normal subjects, four had short REM latency; 65% of normal subjects had mean REM latencies ≥70.0 minutes.
The Mantel-Haenszel (
27) test was used to accommodate within-family correlations of sleep measures and differential numbers of relatives per family. This method takes advantage of a matched-group design, permits estimation of the common odds ratio, and tests the overall significance of the association on the basis of a weighted average of the differences between proportions. Thus, this procedure controlled for within-family correlations and for different numbers of relatives per family. Analysis of variance (ANOVA) and chi-square, linear regression, and logistic regression analyses were also used, depending on questions. When the number of degrees of freedom for analyses is unspecified, df=1.
Thirteen relatives met lifetime criteria for alcoholism. Two relatives consented only to interview, and two used alcohol during the study. Although the remaining nine subjects had been in an extensive period of remission, the time course for the effects of alcohol abuse on polysomnography data is unknown, and these data were excluded. Polysomnographic data of three additional relatives were excluded because of a documented sleep disorder or a violation of the 14-day prestudy preparation period. Clinical data were retained for these 16 relatives in order to test the familial association of major depression.
RESULTS
Chronology of Study
The project was initiated in 1985 at the University of Texas Southwestern Medical Center at Dallas in collaboration with the third and fourth authors (A.J.R., H.P.R.). In November 1986, the first author (D.E.G.) moved to the University of Pittsburgh and began collaboration with the second author (D.J.K.). In 1995, the follow-up phase of the study was moved to the University of Rochester.
Recruitment of the identified participants occurred from 1985 to 1993 in Dallas and Pittsburgh. Approximately 50 subjects were enrolled each year; primary years of recruitment were 1986 to 1992. The first author conducted all initial clinical research diagnostic evaluations and was blind to polysomnography data and to family of origin. Thus, the application of diagnostic criteria and the interviewer were constant, and consistent, across settings. Other clinical interviewers conducted prospective follow-up assessments and were blind to polysomnography data, family of origin, and previous psychiatric history. Polysomnographic records from the University of Texas were rescored at the University of Pittsburgh to standardize scoring (
26) for all records.
Demographic and Clinical Data
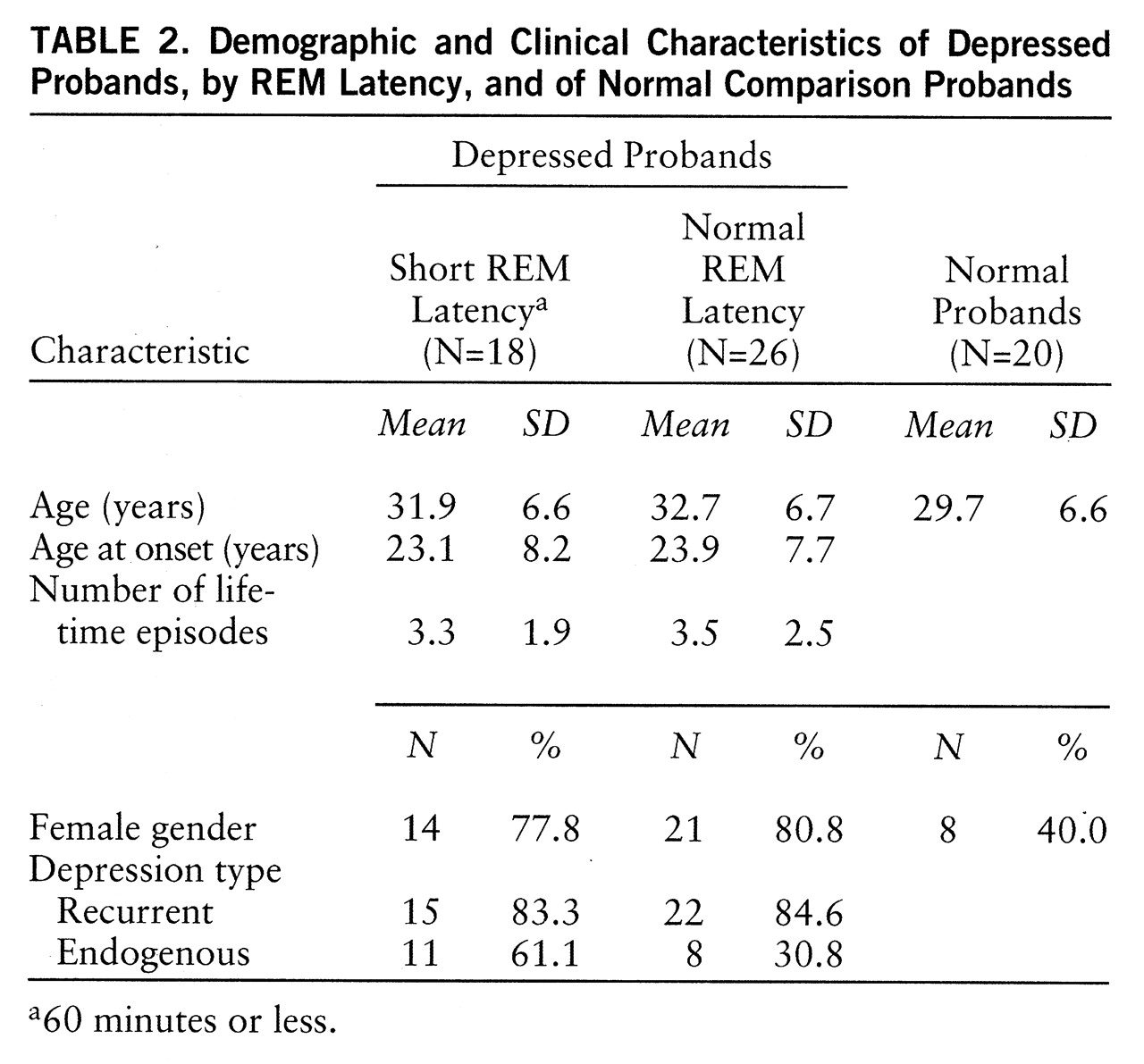
Probands. Clinical data for proband groups were analyzed by using ANOVA or chi-square as appropriate and are presented in
table 2. Probands were similar in age (F=1.16, df=2,63, p=0.32), and the proportion of women did not differ between depressed groups (χ
2=0.06, p=0.81). Female depressed probands outnumbered female normal probands (χ
2=9.8, p=0.007). Age at onset (F=0.11, df=1,43, p=0.74), mean episode number (F=0.12, df=1,41, p=0.73), and proportion with recurrent depression were similar for the two depressed groups. Probands with short REM latency tended more often to have endogenous depression according to RDC (χ
2=3.37, p=0.07). According to criteria, normal probands had no history of psychiatric disorders.
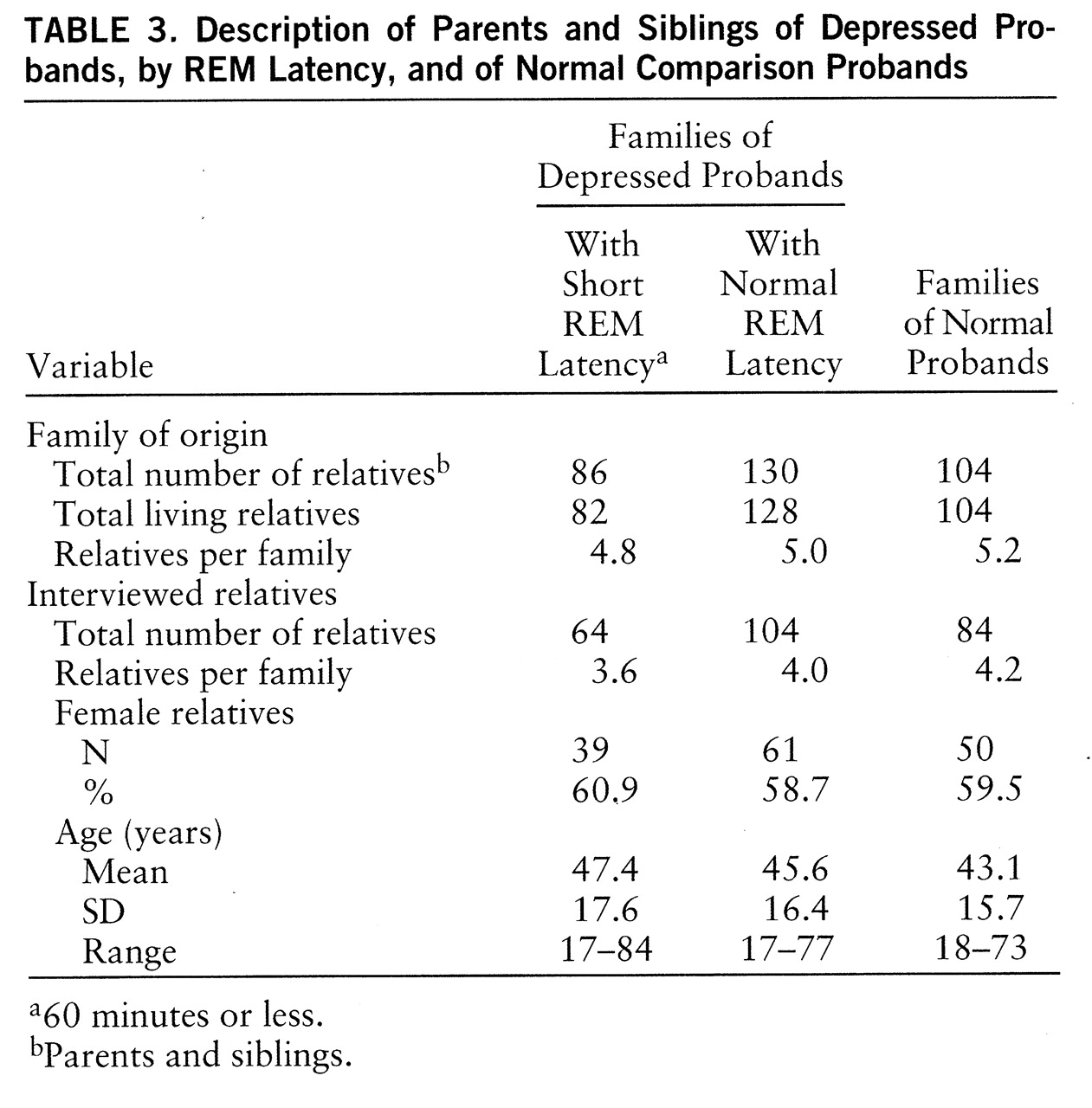
First-degree relatives. Clinical data for 252 parents and siblings were obtained: 168 parents and siblings from depressed probands and 84 parents and siblings from normal comparison probands. One or more siblings were studied in 96% of cases, and two or more siblings were studied in 83% of cases. Polysomnographic data were obtained for 216 relatives (85.7%). Comparative data were analyzed by ANOVA or chi-square, as appropriate, and are presented in
table 3. The number of interviewed relatives per family did not differ (χ
2=0.35, df=2, p=0.84), and sex distribution was closely matched (χ
2=0.09, df=2, p=0.96). Mean ages did not differ (F=1.29, df=2,251, p=0.28). Hamilton depression scale scores were within normal limits in all three groups of relatives (mean=4.3, SD=5.0, versus mean=3.4, SD=3.8, versus mean=1.3, SD=1.5, respectively). As expected, Hamilton depression scale scores of relatives of normal subjects were lower than those of relatives of depressed subjects (F=12.13, df=2,251, p<0.01).
In summary, probands and relatives were comparable on features important to manifestation of depression. Probands from each group were not different in age. Age at onset, number of lifetime episodes, and proportion with recurrent depression were similar between depressed probands with and without short REM latency. Relatives in each group were comparable in size of family, proportion interviewed, proportion studied in the laboratory, mean age, and age range. There were more female depressed than normal probands, consistent with greater prevalence of depression in women. The proportion of female relatives in each group did not differ.
Tests of Hypotheses
Short REM latency is more frequent in relatives of depressed probands with short REM latency. To perform the Mantel-Haenszel (
27) procedure, depressed probands with normal REM latency and normal probands were matched to depressed probands with short REM latency on age, number of siblings, and proportion of female siblings. Matching was done in a manner blind to polysomnography data and diagnoses in relatives. There were, therefore, 18 families of depressed probands with short REM latency matched with 18 families of depressed probands with normal REM latency and 18 families of normal probands.
Odds ratio estimates indicated that the odds of short REM latency were 4.4 times higher in relatives of depressed probands with short REM latency than in relatives of depressed probands with normal REM latency (95% confidence limits=1.39–7.35; χ2=10.15, p=0.001) and 4.3 times higher than in relatives of normal probands (95% confidence limits=1.27–6.66; χ2=10.25, p=0.001). The odds ratio estimate (0.8) for short REM latency in relatives of the two comparison groups was not significant (95% confidence limits=0.35–2.03; χ2=0.10, p=0.75).
Mean REM latencies were significantly shorter in relatives of depressed probands with short REM latency (mean=63.9 minutes, SD=18.7) than in relatives of depressed probands with normal REM latency (mean=75.5 minutes, SD=21.8) and of normal probands (mean=77.5 minutes, SD=27.4) (F=4.19, df=2,21, p=0.02). The last two did not differ from each other. When evaluated as a threshold, short REM latency occurred 1.9 to 2.0 times more frequently among relatives of depressed probands with short REM latency (54.2%) than in relatives of depressed probands with normal REM latency (26.6%) and of normal probands (29.2%) (χ2=15.68, p=0.001).
Major depression is more frequent in relatives of depressed probands with short REM latency. To control for the familial nature of depression, the Mantel-Haenszel procedure was again used. Only data from relatives of depressed probands were included, since normal subjects were explicitly screened for family history of depression.
The odds ratio estimate indicated that the odds of lifetime major depression were 1.5 times higher in relatives of probands with short REM latency than in relatives of probands with normal REM latency (95% confidence limits=1.25–3.03; χ2=4.33, p=0.04). Almost half of the relatives (48.4%, N=31 of 64) of probands with short REM latency reported a history of depression, while only one-quarter of relatives (27.8%, N=29 of 104) of depressed probands with normal REM latency had a history of depression. The relative risk of depression in family members of probands with short REM latency was almost doubled (1.8).
Major depression is more frequent in relatives with short REM latency. The preceding analysis assessed the risk of depression in relatives on the basis of proband REM latency. To evaluate the risk based on relatives' own sleep, we assigned relatives to categories of short and normal REM latency. The lifetime rate of depression among relatives with short REM latency was 50.9% (N=27 of 53). Among relatives with normal REM latency, the rate was only 28.2% (N=24 of 85). The relative risk of depression, based on short REM latency in relatives, was 1.8 (χ2=7.21, p<0.01).
In summary, our hypotheses of both greater frequency of short REM latency and higher rates of depression among relatives of depressed probands with short REM latency were confirmed. Moreover, short REM latency in relatives significantly increased their risk of major depression.
Polysomnographic Profiles of Relatives and Probability of Lifetime Depression
We compared polysomnographic profiles, including REM latency as a continuous variable, in relatives defined by proband types. We then determined the association between lifetime depression and sleep measures.
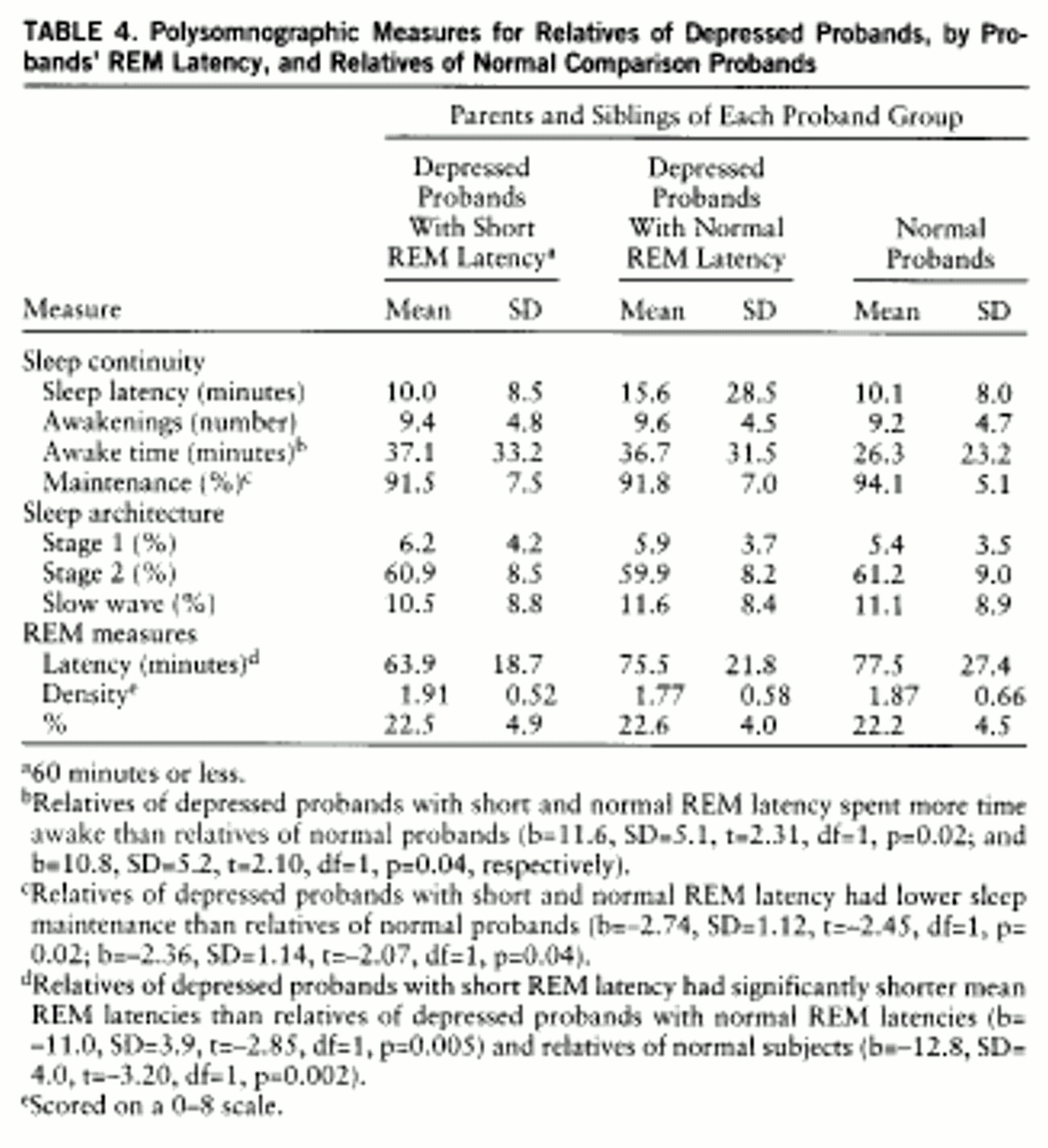
Polysomnographic profiles in relatives. Means and standard deviations of polysomnographic measures for relatives are presented in
table 4. Matched groups, as described earlier, with regression analysis (df=18,116) with dummy variables to accommodate within-family correlations, were used. Modest differences existed in sleep continuity measures between relatives of depressed and normal probands. Relatives of depressed probands with short and normal REM latency spent more time awake (p=0.02 and p=0.04, respectively) and had lower sleep maintenance (p=0.02 and p=0.04) than normal subjects; relatives of depressed groups were not different (b=1.60, SD=6.03, t=–0.26, p=0.79; b=–0.51, SD=1.34, t=–0.38, p=0.70, respectively). Relatives of depressed probands with short REM latency had lower mean REM latencies than relatives of depressed probands with normal REM latency (p=0.005) and normal probands (p=0.002). Relatives of the two comparison groups did not differ (b=–1.9, SD=4.1, t=–0.46, p=0.65).
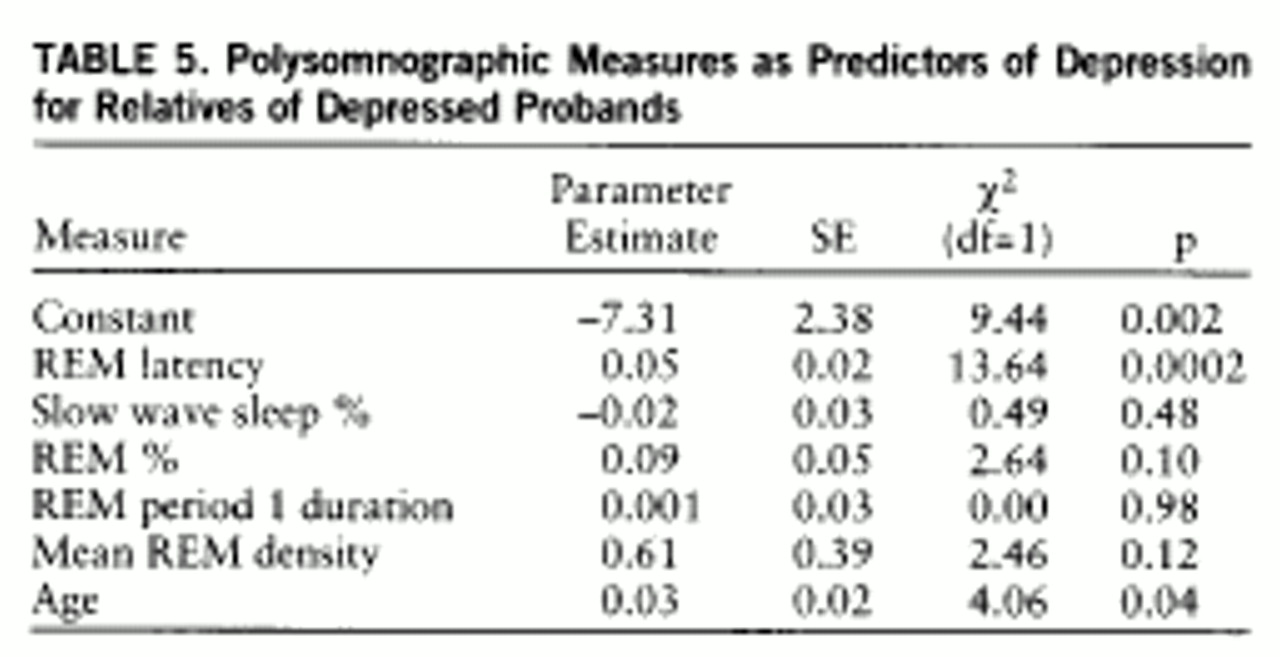
Sleep measures and lifetime depression. To determine whether other polysomnographic measures were associated with lifetime depression in short REM latency and normal REM latency depressed families, we evaluated the constellation of key abnormalities, including slow wave sleep (%), REM sleep (%), length of the first REM period, mean REM density, and REM latency, adjusting for age and using logistic regression analysis in a multivariate model (
table 5). Short REM latency and age were each associated with major depression. When the effects of age and REM latency were included, no other polysomnographic measures significantly predicted outcome. We evaluated combinations of these measures, one at a time and in all permutations, adjusting for age. When measures were evaluated one at a time, REM latency (b=–0.03, SD=0.01; χ
2=10.12, p=0.002) and length of the first REM period (b=–0.05, SD=0.02; χ
2=4.14, p=0.04) were separately associated with depression. The effect of the first REM period was no longer significant (b=–0.01, SD=0.03; χ
2=0.17, p=0.68) when REM latency was added to the equation (b=–0.03, SD=0.01; χ
2=6.34, p=0.01). REM latency continued to predict depression.
Premorbid Polysomnographic Measures
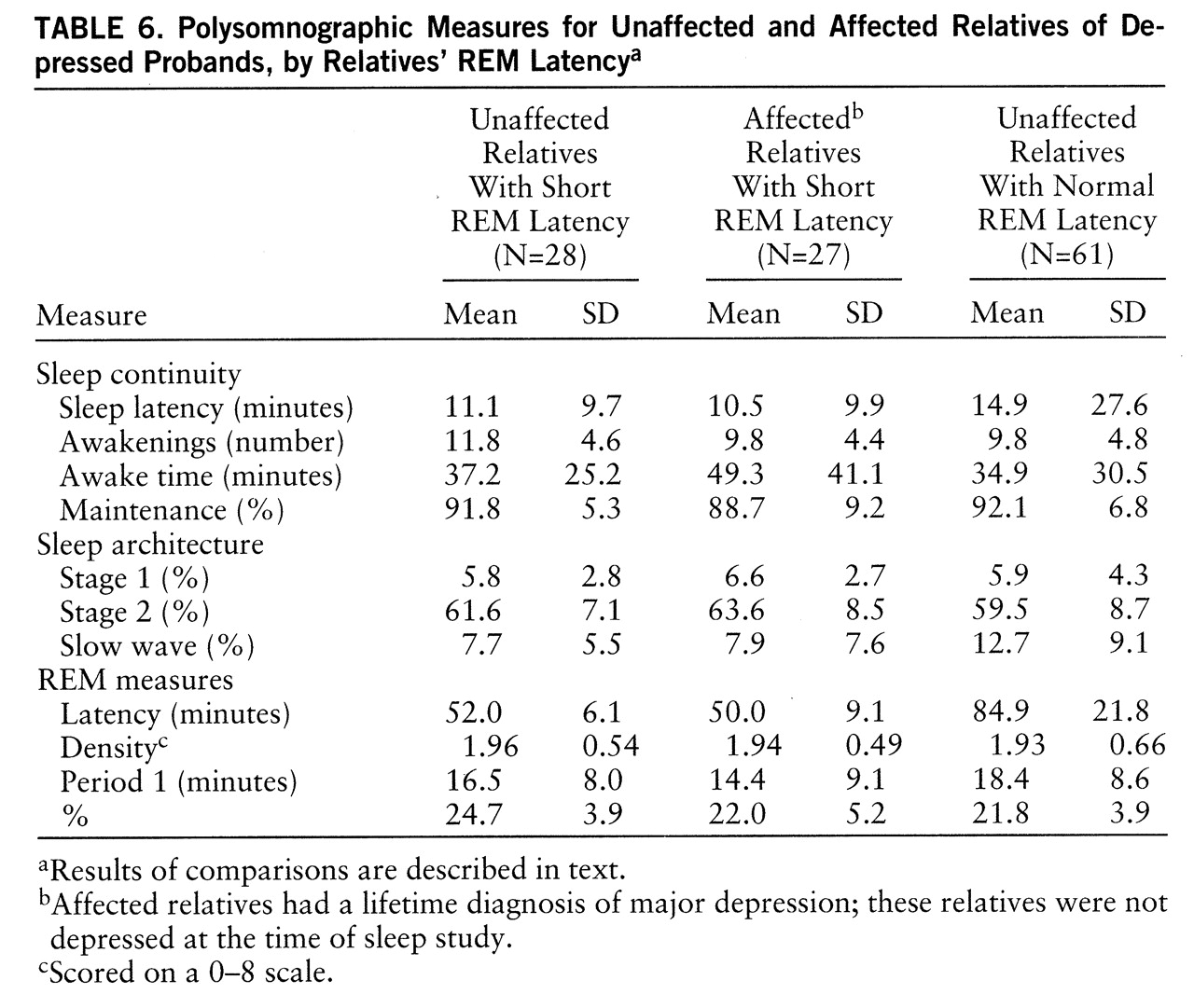
To determine whether other sleep abnormalities were associated with short REM latency in relatives and, further, whether these abnormalities were premorbid, we identified homogeneous groups of short REM latency relatives who were unaffected (i.e., no history of depression) (N=28), short REM latency relatives who were affected (N=27), and relatives of depressed probands with normal REM latency who were unaffected (N=61) (
table 6). Sleep continuity measures did not distinguish the groups (p=0.12 to 0.58). Relatives with short REM latency, whether affected or not, did not differ in slow wave sleep, and both groups had less slow wave sleep than unaffected relatives with normal REM latency (F=8.74, df=2,115, p=0.005). Only REM time and non-REM time distinguished affected and unaffected short REM latency relatives. Unaffected, short REM latency relatives had more time in REM and less in non-REM sleep than either other group (F=7.49, df=2,115, p=0.002, for both measures). These findings are consistent with the position that selected polysomnographic abnormalities occur together and potentially before the first expression of depression.
DISCUSSION
We have a well-characterized cohort of families with substantial evidence of morbidity on the basis of sleep dysregulation. Polysomnographic abnormalities aggregated in families and increased lifetime risk of depression. Specifically, the rate of short REM latency was twice as high in relatives of unipolar probands with short REM latency as in comparison groups (depressed proband families with normal sleep and normal comparison families unselected for polysomnographic abnormalities). Moreover, short REM latency was associated with increased lifetime risk of major depression, whether the abnormality was observed in the proband or in relatives themselves. Short REM latency and slow wave sleep deficits occurred together, yet only short REM latency was associated with increased risk of depression in relatives. We also observed short REM latency, slow wave sleep deficits, and increased REM time in unaffected, at-risk relatives. This profile is consistent with that found in patients with depression and closely mirrors the profile in affected relatives.
One interpretation of these findings is that short REM latency and slow wave sleep deficits are nonspecific, since abnormalities are found in unaffected individuals and in patients with other psychiatric disorders (
8). We suggest, however, that an alternative interpretation is equally viable.
We propose that parallel sleep dysregulation in unaffected and affected relatives is consistent with the position that the dysregulation occurs before the first expression of illness: that is, sleep dysregulation is premorbid. The position that depressive morbidity is related to sleep dysregulation follows from evidence that mechanisms related to sleep abnormalities implicate genetic cosegregation.
The site of the primary sleep defect in unipolar depression is thought to be the first non-REM–REM cycle and includes a prolonged first REM period, increased phasic REM activity, decreased slow wave sleep in the first non-REM period, and short REM latency. These features involve both REM and non-REM sleep regulation. The regulatory defect in slow wave sleep is apparent from loss of the normal exponential decline between the first two non-REM periods. The observation that this blunted decline predicts early recurrence (
28) suggests pathological mechanisms. The regulatory defect in REM sleep is apparent from early onset of the first REM period, redistribution of REM in the sleep period, and increased phasic activity. Pathological implications of REM abnormalities follow from evidence that short REM latency also predicts early recurrence (
29).
The primary sleep defect in depression has been framed in the context of a depressive episode. Interactions between monoaminergic and cholinergic systems link sleep and affective dysregulation. McCarley and Massaquoi (
10) have proposed a neuronal mechanism that accounts for the reciprocal relationship of REM and non-REM sleep dysregulation and the cholinergic supersensitivity found in depressed patients. Weakened monoaminergic inhibition provides the substrate for cholinergic hyperactivity by permitting early onset of cholinergic activity, thereby accounting for cholinergic effects in blocking slow wave activity, shortening the first REM latency, and increasing the intensity and duration of REM sleep. Cholinergic and monoaminergic metabolism and cell counts have been substantiated to be under genetic control, providing a basis for genetic mediation of sleep dysregulation, mood regulation, and depression.
A test for a genetic model requires that abnormalities are present in the asymptomatic state and in unaffected high-risk individuals. Cholinergic supersensitivity has been observed in unaffected at-risk relatives (
30). We also observed preliminary evidence for fundamental modulatory dysregulation in slow wave sleep in our families. Our data suggest that distribution of slow wave sleep was blunted in asymptomatic and unaffected relatives of probands with short REM latency (
31). Of note, whole night slow wave sleep and slow wave sleep in the first two non-REM periods were comparable between relatives of depressed probands with and without short REM latency. Yet, onset of REM distinguished the two groups, suggesting that REM sleep preferentially displaced slow wave sleep. We also have preliminary support for our position that short REM latency confers susceptibility: short REM latency doubled the risk of new-onset depression in a relatively brief prospective follow-up period (mean=3.7 years) (
32).
REM sleep dysregulation has been linked to disturbances in affective information processing (
33,
34). There may be a cognitive/affective mechanism for REM sleep intensification (early onset and increased activation) and subsequent mood dysregulation (
34). Increased phasic activity and inappropriate escape from motor inhibition during REM periods have been suggested as physiological expressions of this affective discharge. Perlis et al. (
34) tested the mood regulatory function of REM sleep and found that mood disturbance was correlated with REM abnormalities, which may interfere with this function. Taken together, these observations implicate premorbid REM sleep dysregulation and subsequent susceptibility to mood dysregulation.
Cosegregation of short REM latency with other polysomnographic abnormalities and with increased frequency of depression is consistent with previous work (
11,
21,
30,
35). In a study of unaffected relatives, Schreiber et al. (
30) did not find baseline REM dysregulation; yet earlier REM sleep onset was documented when a cholinergic probe was used. In another study from the same group, slow wave sleep deficits were also observed (
35). The confluence of slow wave sleep deficits and REM sleep dysregulation supports the contention that sleep parameters, in aggregate, can meaningfully discriminate major depression from other psychiatric conditions (
9,
10).
Finally, our findings suggest marked “familiality” of polysomnographic measures. Although the frequency of short REM latency among relatives of probands with short REM latency is greater, the frequency of short REM latency was 20%–23.9% in the normal comparison group. This finding in normal subjects underscores the familial influence on sleep and weakens the notion that a single polysomnographic measure constitutes a definitive marker for depression. The substantial difference in short REM latency between case and comparison suggests that REM latency is an important marker but cannot stand alone. Polysomnographic abnormalities must be considered in the context of psychiatric history in family of origin. Factors that also influence onset of depression include other measures of sleep, stressful life events, and nonsleep familial factors. Identification and prospective follow-up of at-risk individuals are crucial to documenting the physiological factors that precede the first expression of major depression and provide more direct tests of etiological influences. Determination of risk factors that are differentially related to onset and morbidity in the course of depression as a function of family of origin is a promising focus of ongoing research. Prospective documentation of the clinical course of susceptible individuals can estimate the cumulative weight of these etiologically relevant factors.