It has been suggested that the combination of a high affinity for serotonin 5-HT
2A receptors and a relatively low affinity for dopamine D
2 receptors distinguishes clozapine from typical antipsychotic drugs and accounts for the atypical clinical properties of clozapine (
1). Consistent with this, a high level of 5-HT
2A receptor occupancy (84%–94%) (
2) and a striatal D
2 receptor blockade (38%–63%) which is significantly lower than that reported with typical antipsychotic drugs (70%–89%) (
3) have been recently reported in clozapine-treated schizophrenic patients. However, data concerning the degree of 5-HT
2A occupancy induced by typical antipsychotic drugs in patients are limited.
Using positron emission tomography (PET) and [
18F]~setoperone, a recently developed radioligand with a high affinity for the cortical 5-HT
2A receptor (
4), we examined the binding to this receptor of three antipsychotics in schizophrenic patients. This study focused on chlorpro~mazine, a prototypical neuroleptic with a high in vitro affinity for the 5-HT
2A receptor (
1,
5), in order to determine the degree of its binding to this receptor under clinical conditions. A few clozapine-treated patients were also examined for comparison with chlorpromazine treatment. Also, a few patients treated with amisulpride, a substituted benzamide highly selective for D
2 and D
3 receptors (
6,
7), were examined in order to confirm in vivo this drug's lack of interaction with the 5-HT
2A receptor.
METHOD
The patients in the study met the DSM-III-R criteria for schizophrenia or schizophreniform disorder. The exclusion criteria were organic brain disorder, alcohol or drug abuse, pregnancy, and treatment with a drug, other than the studied drugs, that interacts directly with 5-HT2A receptors. Concomitant medication with benzodiazepines or anticholinergic drugs was allowed. Patients were examined by magnetic resonance imaging to rule out gross cerebral anatomic abnormalities. No treatment was given the morning of the PET studies, which were performed between 9:00 and 11:00 a.m. The protocol was approved by the ethics committee of the Henri Mondor hospital, and patients gave their written informed consent.
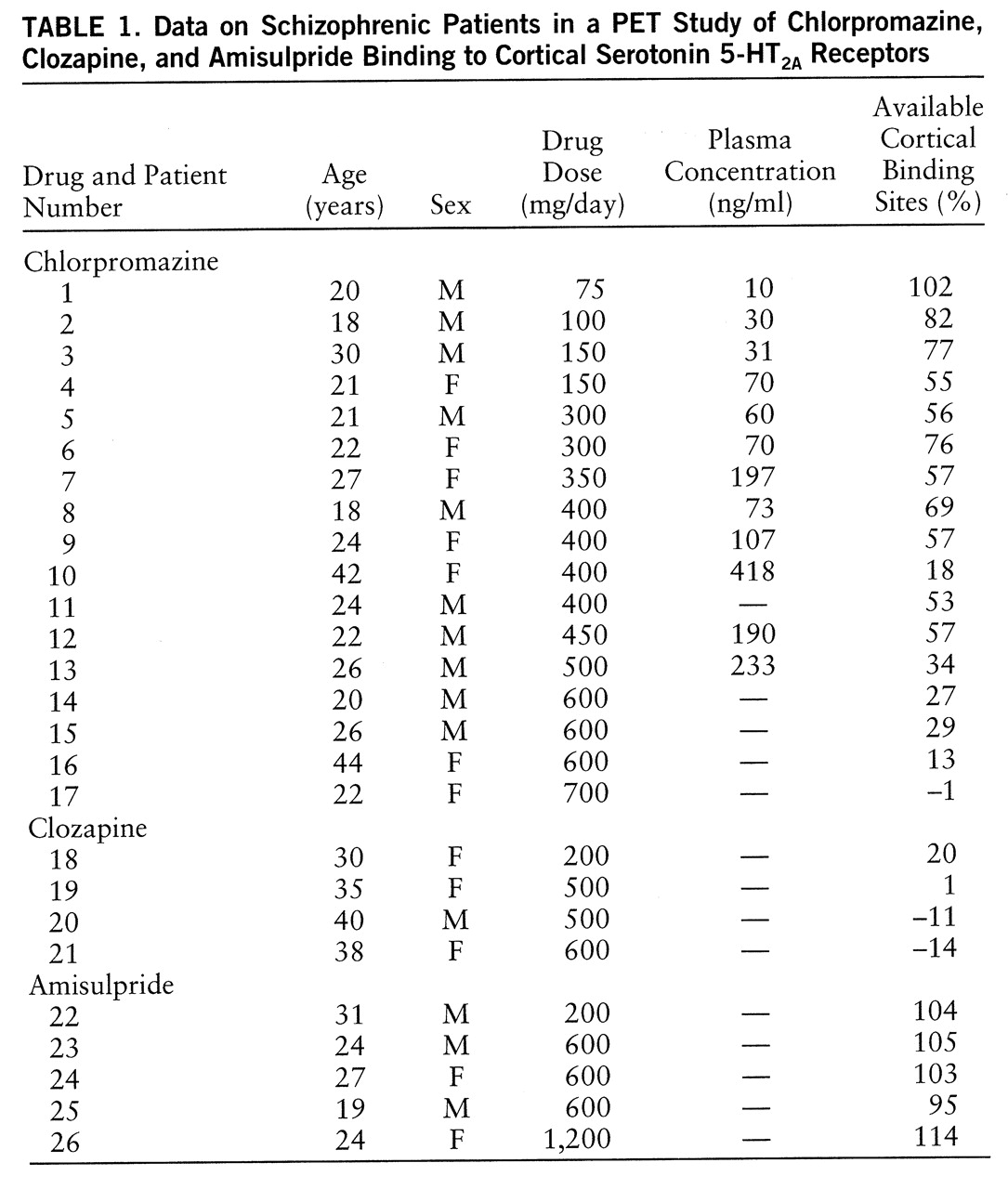
Seventeen chlorpromazine-treated, four clozapine-treated, and five amisulpride-treated patients were recruited according to the treatment prescribed by their physicians (
table 1). The clozapine patients were stable outpatients whose mean length of treatment at the time of the PET scans was 16 months (SD=3, range=12–20). All of the other subjects were inpatients, most of them hospitalized for a psychotic relapse. The mean length of chlorpromazine treatment was 17 days (SD=14, range=5–60), except for one patient who had been treated for 9 months. For the amisulpride group, the mean length of treatment was 49 days (SD=53, range=15–140). To investigate the relation between chlorpromazine dose and degree of 5-HT
2A binding, we selected patients whose chlorpromazine doses varied widely (75–700 mg/day).
Fourteen untreated schizophrenic patients (four women and 10 men; mean age=27 years, SD=9, range=18–44) were used as a control group. Eight had never received an antipsychotic, and six had been free of antipsychotics for at least 5 weeks (range=5 weeks to 3 years), according to interviews with the patients and their relatives and hospital reports.
The PET studies were performed with the LETI TTV01 time-of-flight positron camera as previously described (
4). Briefly, seven cerebral slices parallel to the canthomeatal line were obtained, with the cerebellum on the lowest plane (canthomeatal line plus 10 mm), the striatum on the third plane (canthomeatal line plus 40 mm), and cortical regions on the second to the sixth planes. PET scanning lasted 70 minutes, with image sequences of, successively, 2 minutes (N=5), 5 minutes (N=4), and 10 minutes (N=4). The mean injected doses of radioactivity were 7.09 mCi (SD=1.24) for the reference group, 7.80 mCi (SD=1.70) for the chlorpromazine group, 7.14 mCi (SD=4.27) for the clozapine group, and 8.30 mCi (SD=4.27) for the amisulpride group, with no significant difference among the four groups (F=0.71, df=3,36, p=0.55, one-way analysis of variance [ANOVA]).
During each PET study, a blood sample was drawn from an antecubital venous line 40 minutes after radioligand injection. The ratio between deproteinated-plasma and whole-blood radioactivity concentrations, which has been shown to reflect [
18F]setoperone metabolism (
8), was measured.
For 12 of the chlorpromazine-treated patients, plasma chlorpromazine concentrations were also measured from samples drawn just before their PET scans. Samples were frozen at –80°C and afterward analyzed together by routine radioimmunoassay.
Cortical and cerebellar regions of interest were determined in all patients by means of a standardized protocol. External brain boundaries were first delimited on three planes (canthomeatal plus 40 mm to canthomeatal plus 70 mm) with the use of a 40% radioactivity isocontour on one early PET image (4–6 minutes). The cortex was then operationally defined as an 8-pixel-thick ring within this isocontour. A region of interest was drawn on each cerebellar hemisphere in the canthomeatal plus 10 mm plane. Thereafter, cortical and cerebellar regions of interest were transferred onto all of the PET images obtained in the same plane. The cortical radioactivity concentration was defined as the mean value for cortical regions of interest in the three planes, and the cerebellar radioactivity concentration as the mean for the right and left cerebellar regions of interest. To normalize for intersubject variations in the amount of injected radioactivity, the concentration values are expressed as the percentage of injected activity per liter of brain tissue.
PET Determination of Antipsychotic Drug Binding to 5-HT2A Receptors
We used the reduction in the number of sites available for the binding of [
18F]setoperone in the cortex, a region where it has been demonstrated that [
18F]setoperone selectively labels the 5-HT
2A receptor (
4), to estimate the binding of the studied antipsychotics to this receptor.
The cortical binding potential of the radioligand was first calculated for each patient. Binding potential equals the product of receptor density and affinity and therefore reflects the capacity of a tissue to specifically interact with a ligand (
9). Assuming tracer conditions, binding potential can also be expressed as the ratio at equilibrium between the specifically bound and the free and nonspecifically bound radioligand concentrations (
9). In this study, nonspecifically bound radioligand concentration was estimated by the concentration of radioactivity in the cerebellum, a region where specific binding of the radioligand is negligible (
4), and specifically bound radioligand concentration was estimated by the difference between cortical and cerebellar radioactivity concentrations. The binding potential was calculated as the ratio of specifically bound to nonspecifically bound radioligand concentrations at 45 minutes (i.e., from the data of the 40- to 50-minute PET image), since it was the mean time of peak value for specifically bound radioligand concentration in the patients and in the control subjects, indicating that an apparent equilibrium was reached.
The percent reduction of binding potential in the treated patients with respect to the binding potential estimated in the absence of drug treatment (baseline) was then used to estimate the percentage of available binding sites for the radioligand, according to the following equation: percentage of available binding sites equals the binding potential in treated patients divided by the estimated binding potential at baseline, multiplied by 100. The baseline binding potential was defined for each treated patient as the binding potential value expected in an untreated patient of the same age, according to the linear regression found in the reference group (binding potential=2.0670– 0.0294×age [years]) (r=0.90, N=14, p=0.0001). The reduction of binding potential with age observed here is consistent with the age-related reduction in cortical 5-HT
2A receptor density previously reported in vivo in control subjects (
4,
10) and in postmortem studies (
11,
12).
Animal studies have shown that both clozapine and chlorpromazine induce a significant 5-HT
2A receptor down-regulation in the frontal cortex (
13,
14). It is not yet known whether these drugs produce a similar effect in humans. Since the PET procedure used in this study did not allow us to differentiate the decrease in cortical binding potential attributable to occupancy and to down-regulation, we expressed the results as percentages of available binding sites for the radioligand, rather than as receptor occupancy percentages.
Statistical Analysis
Comparisons of the four groups of patients were conducted with the use of one-way ANOVA. The relation between the oral dose or plasma concentration of chlorpromazine and the percentage of available binding sites was tested with the Spearman rank correlation coefficient.
RESULTS
No significant differences in [
18F]setoperone radioactivity concentration in the cerebellum were observed among the reference group (mean=1.06% of injected activity per liter of brain tissue, SD=0.31%), the chlorpromazine group (mean=0.91%, SD=0.23%), the clozapine group (mean=1.03%, SD=0.21%), and the amisulpride group (mean=0.87% SD=0.12%) (F=1.25, df=3,36, p=0.31). This finding supports the hypothesis that the specific binding of [
18F]~setoperone in the cerebellum is negligible in the conditions of this study (
4). Similarly, the four groups of patients did not differ in the ratio of deproteinated-plasma to whole-blood radioactivity concentrations (mean=0.30, SD=0.13; mean=0.35, SD=0.13; mean=0.28, SD=0.06; and mean=0.26, SD=0.05, respectively; F=1.08, df=3,36, p=0.37), suggesting no difference in [
18F]setoperone metabolism.
The mean percentage of available cortical binding sites was 51% (SD=27%) in the chlorpromazine group (
table 1). This percentage was negatively correlated with the oral dose of chlorpromazine (r=–0.81, N=17, p=0.001) and with its plasma concentration (r=–0.78, N=12, p=0.01). The mean percentages of available binding sites were –1% (SD=15%) and 105% (SD=7%) in the clozapine and amisulpride groups, respectively.
DISCUSSION
Using PET in chlorpromazine-treated schizophrenic patients, we found a dose- and plasma concentration-dependent reduction in available cortical binding sites for [18F]setoperone, a high affinity 5-HT2A receptor antagonist. This provides evidence for a significant binding of chlorpromazine to cortical 5-HT2A receptors under clinical conditions. For a daily dose of 400 mg, the mean reduction in available cortical binding sites was about 50%, while for the patient receiving the highest dose (700 mg/day), the percentage of available binding sites was close to 0%, suggesting a virtual saturation of 5-HT2A receptors.
Consistent with the high level of 5-HT
2A occupancy reported previously with clozapine (
2), a very low percentage of available cortical binding sites was observed in all of the patients in this study who were treated with this drug. The patient receiving the lowest dose (200 mg/day) had only 20% of available binding sites, while for the highest doses (500–600 mg/day) percentages close to 0% were observed. This confirms that therapeutic doses of clozapine are regularly associated with at least an 80% blockade of 5-HT
2A receptors (
2). For the amisulpride-treated patients, the mean percentage of available cortical binding sites was close to 100%. This supports the lack of significant in vivo interaction of amisulpride with the 5-HT
2A receptor, which is consistent with the in vitro binding profile of this drug.
Since the benefit of clozapine over chlorpromazine has been demonstrated in several clinical trials (
15,
16), the results of the present study can be viewed in the context of hypotheses for the atypical clinical properties of clozapine. We observed a very high degree of 5-HT
2A receptor blockade with clozapine at all of the studied doses. A main finding of this study, however, was that a virtual saturation of the 5-HT
2A receptors can also be induced by high doses of chlorpromazine. This highlights the importance of taking into account the administered doses to compare the in vivo biological effects of both drugs in patients. For instance, in the seminal study by Kane et al. (
15), the superiority of clozapine over chlorpromazine in schizophrenic patients resistant to typical antipsychotic drugs was demonstrated by using a drug regimen of 400–600 mg/day for clozapine and more than 1,000 mg/day for chlorpromazine. Our results suggest that at these dose levels, a virtual saturation of the 5-HT
2A receptors was likely in both groups of patients and could not account per se for the demonstrated difference in the therapeutic efficacy of the two drugs.
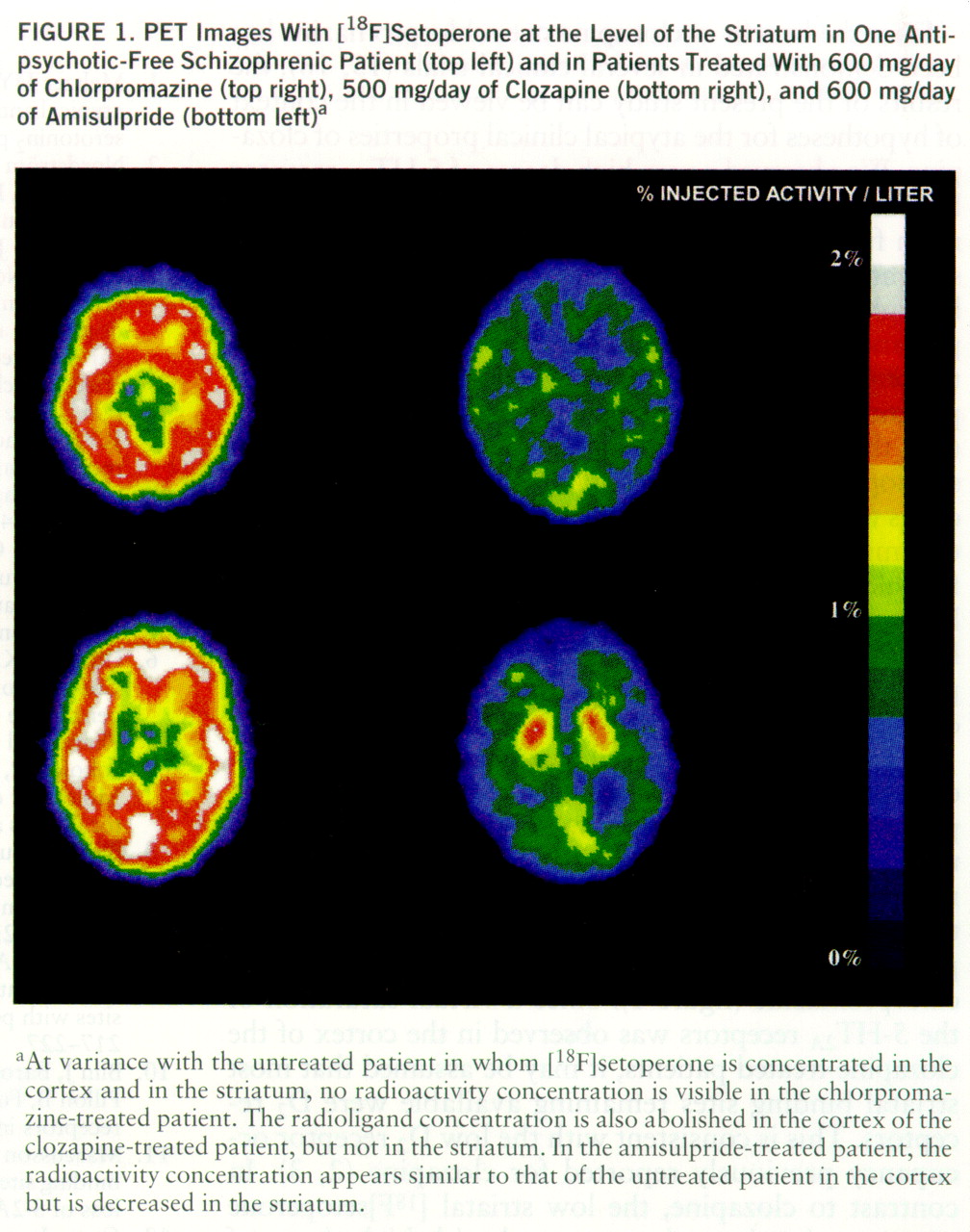
In this study, we observed that the most visible difference on PET images between the clozapine- and chlorpromazine-treated patients was in the striatum, a structure where [
18F]setoperone binds to both D
2 and 5-HT
2A receptors (
4). Indeed, a striatal [
18F]setoperone concentration was clearly visible in all of the clozapine-treated patients but not in the patients receiving high doses of chlorpromazine (
figure 1). Since a virtual saturation of the 5-HT
2A receptors was observed in the cortex of the clozapine-treated patients, it may be assumed that most striatal binding sites remaining available were D
2 receptors. This is consistent with the low D
2 receptor occupancy previously reported for clozapine (
2,
3). In contrast to clozapine, the low striatal [
18F]setoperone concentration in patients treated with high doses of chlorpromazine suggests a high level of blockade of both 5-HT
2A and D
2 receptors. These findings are consistent with the hypothesis that a relatively higher ratio of occupancy of 5-HT
2A versus D
2 receptors distinguishes in vivo clozapine from typical antipsychotics (
1). It may be emphasized, however, that the difference reported here between clozapine and chlorpromazine mainly rests on the low degree of D
2 receptor occupancy induced by conventional doses of clozapine.
This different degree of D
2 receptor occupancy may account for differences in clinical properties. Specifically, the failure of clozapine to produce a high striatal D
2 receptor blockade at conventional doses is sufficient per se to account for the low incidence of extrapyramidal side effects associated with this treatment (
3). Also, one of the factors relevant to the suggested action of clozapine on negative schizophrenic symptoms (
17) may be a low striatal D
2 occupancy. Indeed, amisulpride was recently reported to improve negative symptoms (
18,
19) at doses associated with a low striatal D
2 receptor occupancy (
20). Thus, a low degree of striatal D
2 receptor blockade may be a condition for an antipsychotic drug to be able to improve negative schizophrenic symptoms independent of any action on the 5-HT
2A receptor.