Cocaine's binding to the dopamine transporter is the mechanism most widely implicated in its addictive potential (
1,
2). Less clearly understood, however, is the degree to which altered dopamine transporter regulation is a factor in the pathophysiology of cocaine abuse. A view of dopamine transporter regulation in human cocaine-abusing subjects may improve our understanding of clinical aspects of cocaine dependence, including drug-induced craving, dysphoria, and relapse.
A considerable, albeit inconsistent, body of research has examined the effects of cocaine administration on dopamine transporter regulation. Homogenate binding studies have documented increased, decreased, and unchanged brain densities (B
max) of dopamine transporters in laboratory animals exposed to cocaine (
3,
4). In contrast, several (
5,
6), but not all (
7), postmortem studies of human cocaine-related deaths have noted dramatic increases (50%–500%) in dopamine transporter binding sites as measured by cocaine analogues. On the basis of its structural similarity to cocaine, we have used the iodinated radioligand [
123I]β-carbo~methoxy-3β-(4-iodophenyl)tropane ([
123I]β-CIT; also known as [
123I]RTI-55) to image striatal dopamine transporters in cocaine-abusing subjects with single photon emission computed tomography (SPECT).
METHOD
Twenty-eight cocaine-dependent (by DSM-III-R criteria) and 24 healthy comparison subjects, matched as a group for age (mean=32 years, SD=6, versus mean=33, SD=9, respectively) (t=0.20, df=51, p=0.84, two-tailed unpaired t test) and gender (10 women and 18 men versus nine women and 15 men) (χ2=0.02, df=1, p=0.89), were studied. Cocaine-dependent subjects were frequent (mean=25 days/month, SD=8) and heavy (mean=30 g/month, SD=23) intravenous or freebase (N=27) users of the drug and were free of other primary psychiatric or substance use disorders. Subjects had physical and neurological examinations, ECG, and routine blood and urine laboratory tests to rule out concurrent medical illness and pregnancy, as well as exclude or verify illicit drug use. All provided voluntary written informed consent for study procedures.
Cocaine-abusing subjects were studied during a period of acute (96 hours or less) drug abstinence and remained hospitalized on a locked inpatient research unit for the entire study. Subject assessments included the Cocaine Craving Scale (
8), the Hamilton Depression Rating Scale (
9), and the Hamilton Anxiety Rating Scale (
10). Healthy comparison subjects were studied as outpatients.
Subjects received an injection of [
123I]β-CIT (dose mean=355 MBq, SD=44, or mean=9.6 mCi, SD=1.2; radiochemical purity mean=97.4%, SD=1.9%; specific activity >185 GBq/mmol or >5,000 Ci/mmol) followed approximately 24 hours later by SPECT scanning on the brain-dedicated CERASPECT camera (Digital Scintigraphics, Waltham, Mass.) (
11). Studies in healthy human subjects have demonstrated that [
123I]β-CIT reaches equilibrium binding in the brain by 24 hours (
12), so that a simple unitless ratio of regional radioactivities (V
3″=B
max/K
d/V
2=specific/nondisplaceable binding=[striatum–occipital]/occipital) can be used to estimate dopamine transporter number (i.e., B
max), by assuming comparable dopamine transporter affinities (1/K
d) and nondisplaceable distribution volumes (V
2) in both study populations. Equilibrium assumptions were directly tested by obtaining three SPECT scans in the first 12 cocaine-abusing subjects and comparison subjects at 24, 27, and 30 hours. Before imaging, four Na
99mTcO
4-filled fiducial markers (6–10 µCi) were glued bilaterally along the canthomeatal line to permit identification of this plane during image analysis.
Images were reconstructed from photopeak counts (mean=159 keV, SD=16) by filtered (Butterworth; cutoff=1 cm, power factor=10) back-projection, displayed as a 64×64×32 matrix (voxel size=3.3×3.3×3.3 mm) and reoriented to the canthomeatal plane. The four slices with the highest striatal uptake were summed and attenuation corrected (µ=0.15 cm
–1) to yield a final transaxial slice 13.3 mm thick. Standardized region of interest templates for left and right caudate/putamen and occipital lobe (
13) were visually applied to obtain measures of regional radioactive density, decay corrected to the time of injection.
Statistical differences between groups were assessed by two-tailed unpaired t tests; comparisons of [123I]β-CIT binding and clinical ratings employed Pearson's product-moment correlation. Statistical significance was assumed at the p<0.05 level.
RESULTS
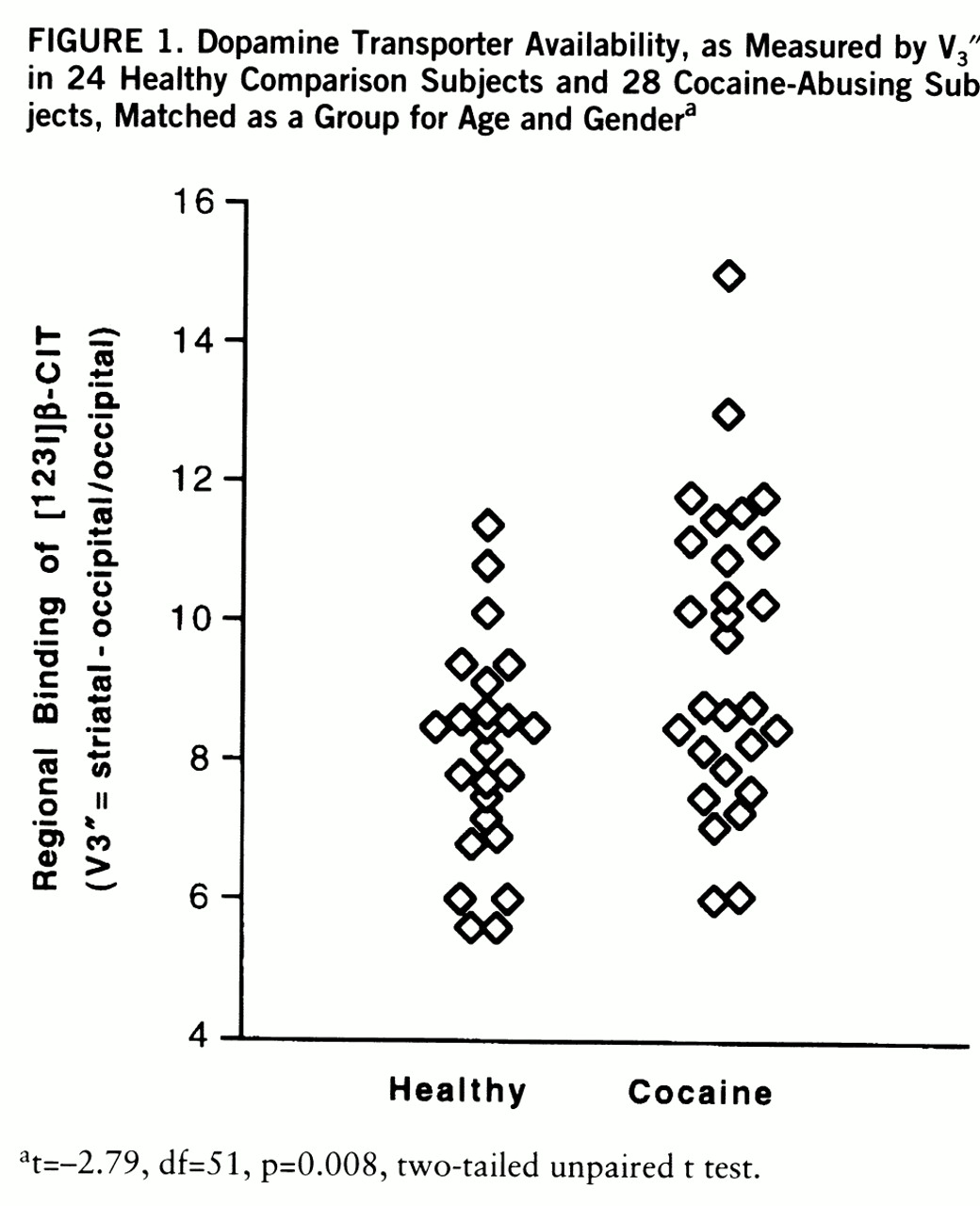
Results revealed modest (approximately 20%), albeit statistically robust (p=0.008), increases in V
3″ in cocaine-abusing subjects as compared to comparison subjects (mean=9.5, SD=2.1, versus mean=8.1, SD=1.5) (
figure 1). Elevations in V
3″ did not derive from differences in nondisplaceable (i.e., occipital) binding between groups (cocaine versus comparison: mean=0.16, SD=0.05×10
–2, versus mean=0.15, SD=0.05×10
–2% ID · ml
–1) (t=–1.07, df=51, p=0.29). Clearance rates for specific (mean=0.17%/hour, SD=0.6, versus mean=0.00%/hour, SD=0.6) (t=0.58, df=23, p=0.58) and nondisplaceable (mean=–1.91%/hour, SD=2.1, versus mean=–0.53%/hour, SD=2.3) (t=–1.73, df=23, p=0.11) binding were comparable for the addicted and nonaddicted groups and were consistent with equilibrium assumptions (
12). Among severity of use and behavioral ratings, only levels of depression (Hamilton depression scale score) were significantly correlated (r=–0.50, df=26, p=0.02) with [
123I]β-CIT binding in cocaine-abusing subjects.
DISCUSSION
The present study provides the first in vivo evidence of increased cocaine binding sites (i.e., striatal dopamine transporters) in cocaine-abusing subjects as measured by [
123I]β-CIT SPECT. Our findings of increased V
3″ in the addicted group are most likely explained by an increased density of dopamine transporters, as suggested by two previous postmortem reports. Little et al. (
5), reporting on seven cocaine-related deaths, found [
3H]~WIN 35,428 binding to be 50%–200% higher than in seven comparison subjects as measured by quantitative autoradiography. Staley et al. (
6) independently confirmed these findings in six cocaine overdose victims, demonstrating selective increases (approximately 500%) in B
max for the high- but not the low-affinity binding site. In contrast to these postmortem studies, we observed more modest (approximately 20%) and overlapping degrees of dopamine transporter elevation in our cocaine-abusing subjects (
5,
6), results that may explain negative findings in other postmortem (
7,
14,
15) and positron emission tomography (PET) (
16) studies.
The pathophysiological significance, if any, of increased dopamine transporters in cocaine-abusing subjects is currently unknown. Specifically, it remains to be established whether increases in cocaine binding sites reflect a premorbid, perhaps predisposing, trait in susceptible individuals, or whether dopamine transporter elevations are a secondary, potentially homeostatic, response to chronic dopamine reuptake blockade by cocaine. Future SPECT studies of individuals at high risk for cocaine addiction or previously addicted individuals during periods of sustained drug abstinence may ultimately help to answer this question. In favor of the former possibility, we found no evidence of a relationship between a variety of severity of use measures and dopamine transporter binding. Conversely, our observation of an association between depressed mood and low dopamine transporter levels provides indirect support for an adaptive response. In the absence of a more detailed understanding of the molecular basis of elevations in [
123I]β-CIT binding, such inferences remain highly speculative. However, our Hamilton depression scale finding is corroborated by previous PET findings of an inverse correlation between depressed mood and striatal D
2 receptors (
17). Such abnormalities in depressed cocaine-abusing subjects are consistent with preclinical research suggesting an important role for dopaminergic dysfunction in postcocaine anhedonia (
18). Thus, prospective replication of this preliminary finding by future investigations will be critical and may ultimately improve our understanding of the neurobiological basis of cocaine-related dysphoria and relapse.