Treatment of negative symptoms remains a challenge in the management of schizophrenia. These symptoms can be the most persistent, intractable, and disabling element of the illness for many patients. Uncertainty surrounds the nature of negative symptoms, the validity of current measures of these symptoms, their responsiveness to pharmacological intervention, and the appropriateness of treatment. First, it is difficult to discriminate between the enduring deficit symptoms of schizophrenia and the less stable negative features arising as a consequence of extrapyramidal side effects of antipsychotic drugs, depressive states, and institutionalization
(1). Second, the question of the usefulness of antipsychotic drugs in the treatment of negative symptoms remains unresolved
(2). A major methodological problem is how to differentiate a direct improvement in primary negative symptoms from an indirect improvement due to relief of positive, extrapyramidal, and depressive symptoms
(3–
8). Clinical trials with recently developed atypical antipsychotics provide only limited information in this regard, since few have been conducted specifically in patients with predominantly primary negative symptoms. Retrospective analyses using path analytical approaches for schizophrenic patients with mixed positive and negative symptoms provide some evidence of a direct effect of atypical antipsychotics on negative symptoms
(9,
10), although definitive proof of drug efficacy for primary negative symptoms remains to be established.
Amisulpride is a substituted benzamide with a unique neurochemical and psychopharmacological profile. It has high selectivity for dopamine D
2 and D
3 receptor subtypes in the limbic region and blocks functional responses mediated by these receptors
(11,
12). It has no appreciable affinity for other receptors. At high doses, amisulpride exhibits dopaminergic blocking activity similar to that induced by classical antipsychotics, while at lower doses it appears to facilitate dopaminergic transmission through preferential blockade of presynaptic dopamine autoreceptors. This combination of pharmacological properties may account for its atypical profile in animal models, where it shows activating and prohedonic properties at low doses and an absence of cataleptogenic effect even at high doses
(13).
This atypical profile may also explain the clinical efficacy of amisulpride against acute psychotic symptoms at high doses and predominant negative symptoms at low doses, as well as its low propensity for causing extrapyramidal symptoms
(14). Amisulpride, 400–1200 mg/day, has demonstrated similar clinical efficacy to that of haloperidol, 15–40 mg/day
(15–
17), and flupenthixol, 15–25 mg/day
(18), in the treatment of patients with acute exacerbations of schizophrenia. At lower doses (mostly 100–300 mg/day), amisulpride was more effective than placebo in well-controlled studies of patients with primary negative symptoms of schizophrenia
(19–
21). The substantial improvement in negative symptoms observed in these studies was not coupled with a change in positive symptoms or parkinsonism, which remained mild during treatment.
The main objective of this study was to compare the efficacy of two low doses of amisulpride (50 and 100 mg/day) with placebo in the treatment of negative symptoms of schizophrenia. The study also assessed the safety of the two active treatments on the basis of clinical and laboratory data.
METHOD
A total of 242 patients meeting the following criteria were included in the study: age between 18 and 60 years and a DSM-III-R diagnosis of schizophrenia, residual type (diagnosis 295.6), of no more than 20 years’ duration (mentally retarded patients were excluded). This maximum duration of illness was chosen to increase the possibility of change, since extremely chronic patients might not be reactive even after 3 months of treatment
(22). The subjects had to present predominantly negative symptoms, with a total score of 60 or more on the Scale for the Assessment of Negative Symptoms (SANS)
(23) and a total score of 50 or less on the Scale for the Assessment of Positive Symptoms (SAPS)
(24). Patients receiving depot antipsychotic therapy were eligible for inclusion in the placebo washout phase only if the time elapsed since the last injection was at least equal to the usual time interval between injections. Patients were hospitalized or ambulatory; compliance was assessed by means of amisulpride plasma assays at the last evaluation as well as counts of unused capsules at each visit.
Exclusion criteria included the following: 1) patients with advanced cardiovascular, renal, hepatic, respiratory, hematological, or endocrinological disease; pheochromocytoma; Parkinson’s disease; alcohol or other substance abuse; or known hypersensitivity or allergy to benzamides; 2) patients in whom discontinuation of antipsychotic, antidepressant, and/or mood-regulating therapy did not seem possible; 3) pregnant women, women of child-bearing potential, and nursing mothers; and 4) patients who had participated in a clinical trial during the previous 6 months.
The study was conducted in accordance with the Helsinki declaration of 1964 and the Tokyo (1975), Venice (1983), and Hong Kong (1989) amendments. The protocol was approved by the local ethics committee of the participating center (or country, if applicable). After complete explanation of the study to the subjects, their written informed consent was obtained.
This multicenter, multinational, double-blind placebo-controlled trial consisted of two successive phases: a 4-week, single-blind washout period in which patients received one placebo capsule daily and a 12-week, double-blind study period in which patients were randomly assigned to receive either amisulpride, 50 or 100 mg/day, or placebo. All drugs were administered orally once daily. Antiparkinson agents were phased out during the first 2 weeks of the washout period. Continuation of hypnotic agents used previously on a regular basis was allowed throughout the washout period, and if necessary, initiation of a hypnotic agent was allowed during the study. Administration of an anxiolytic agent (lorazepam, ≤5 mg/day) for no more than two periods of up to 7 days, separated by at least 7 days, was allowed to control transient anxiety; patients requiring further treatment were withdrawn from the study.
Efficacy and Safety Evaluation
The primary efficacy measure was the change in total SANS score. Secondary efficacy assessments included SANS subscale scores; SANS responders; SAPS score; Brief Psychiatric Rating Scale (BPRS)
(25) score, with 18 items scored from 1 to 7; Montgomery-�sberg Depression Rating Scale
(26) score, with 10 items scored from 0 to 6; and score on the Clinical Global Impression (CGI) scale
(27) with three items.
At each visit, safety was evaluated with the Simpson-Angus Rating Scale
(28) for extrapyramidal manifestations and the Abnormal Involuntary Movement Scale (AIMS)
(29). Blood pressure and heart rate were also evaluated.
All efficacy and safety evaluations were performed on study days 0, 14, 28, 56, and 84 or at the time of a patient’s withdrawal from the study. Routine laboratory tests were performed during the last 2 weeks of the washout period and at study endpoint.
Assessments were performed by the investigators, who had participated in two videotaped training sessions of rating procedures before the study began. Formal calculations of interrater reliability were not performed.
Statistical Analysis
Statistical analyses were done on an intent-to-treat basis, including all randomly assigned patients who had at least one available treatment evaluation. Demographic and baseline characteristics were compared by means of a one-way analysis of variance (ANOVA) (treatment factor) for quantitative variables; when the global treatment effect was significant, the three pairwise comparisons between two treatment groups (amisulpride-50 mg versus placebo, amisulpride-100 mg versus placebo, and amisulpride-50 mg versus amisulpride-100 mg) were performed according to J. Kunert’s method
(30). This method ensures that each of the three pairwise comparisons can be done with an alpha risk equaling 0.05. For the efficacy and safety criteria, an ANOVA was carried out for the quantitative variables (a two-way analysis [factor treatment and country] for the main efficacy criteria—SANS scores—and a one-way analysis for all other parameters). Fisher’s exact test or chi-square tests were used for dichotomous variables.
In addition to the intent-to-treat analysis, an analysis of data from the efficacy assessment scales of the patients who completed the last study evaluation (completers) was conducted with the use of the tests listed above.
RESULTS
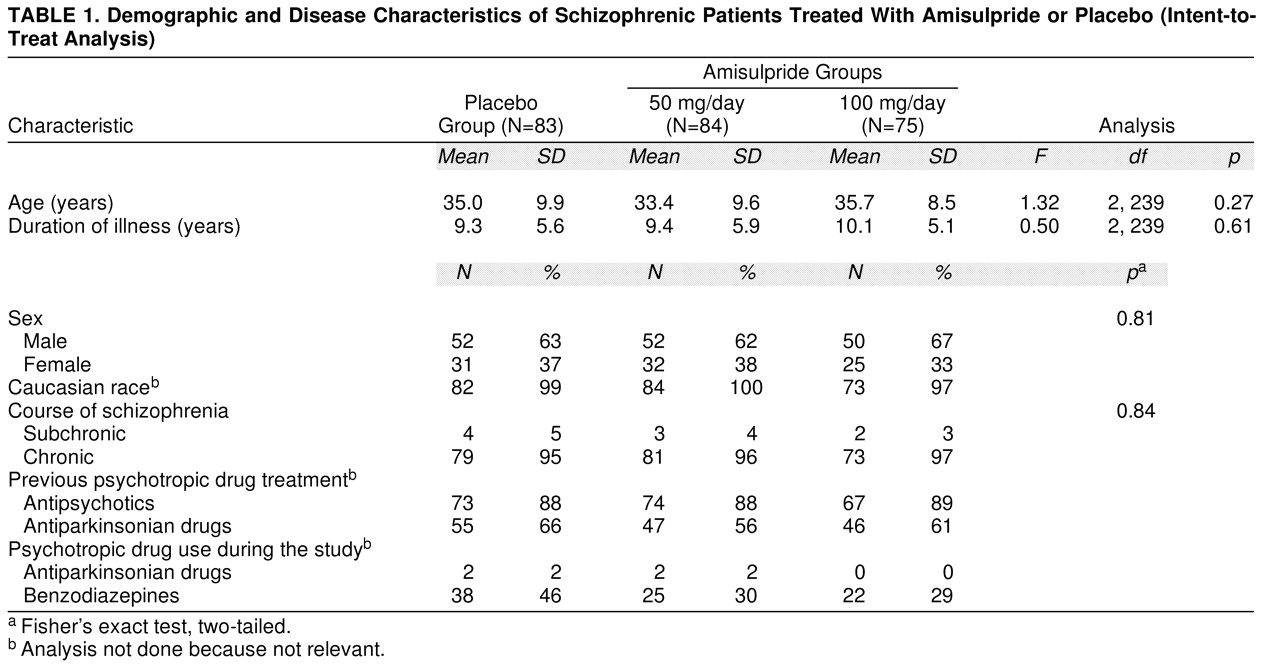
Between January 1993 and July 1996, a total of 242 patients (154 men, 64%, and 88 women, 36%) in 35 centers in four countries were randomly assigned to treatment. Their mean age was 34.7 years (SD=9.4). The three treatment groups were comparable in terms of demographic and disease characteristics at baseline (
table 1). In the 6 months preceding study entry, most patients (88%) had received antipsychotic medication; five, nine, and six patients in the placebo, amisulpride-50 mg, and amisulpride-100 mg groups, respectively, had received a depot neuroleptic 4 weeks prior to washout. During the study, nearly one-third of the patients received at least one psychotropic drug other than an antipsychotic.
Efficacy
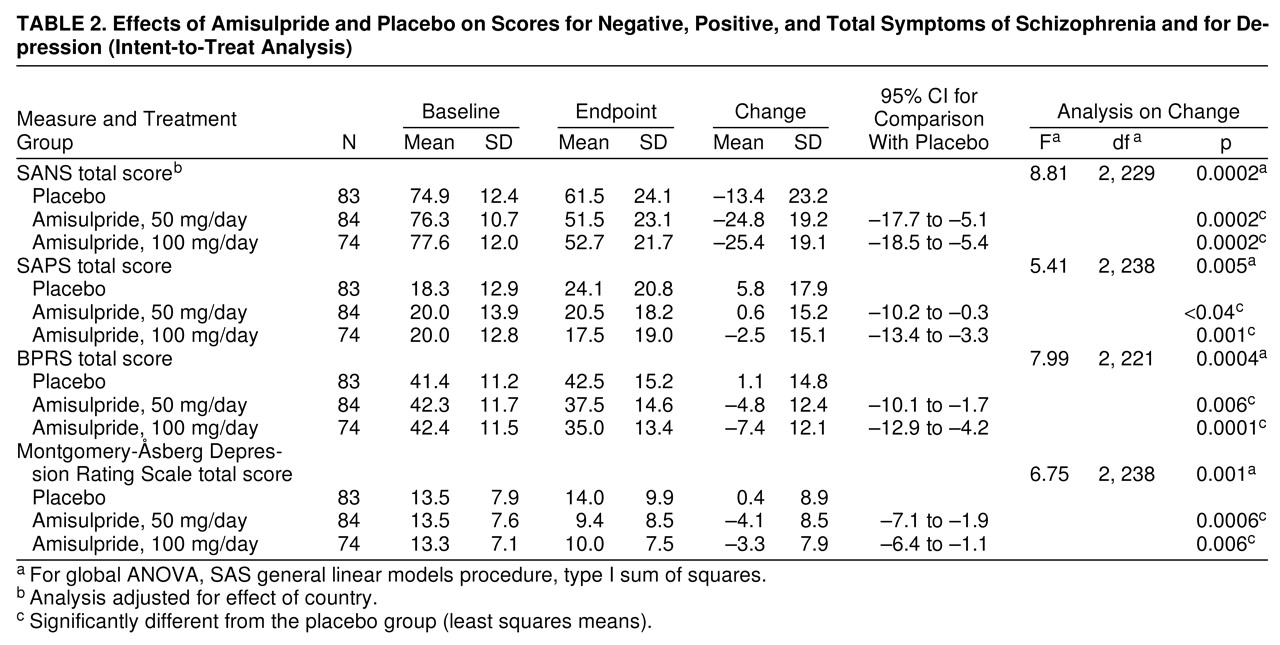
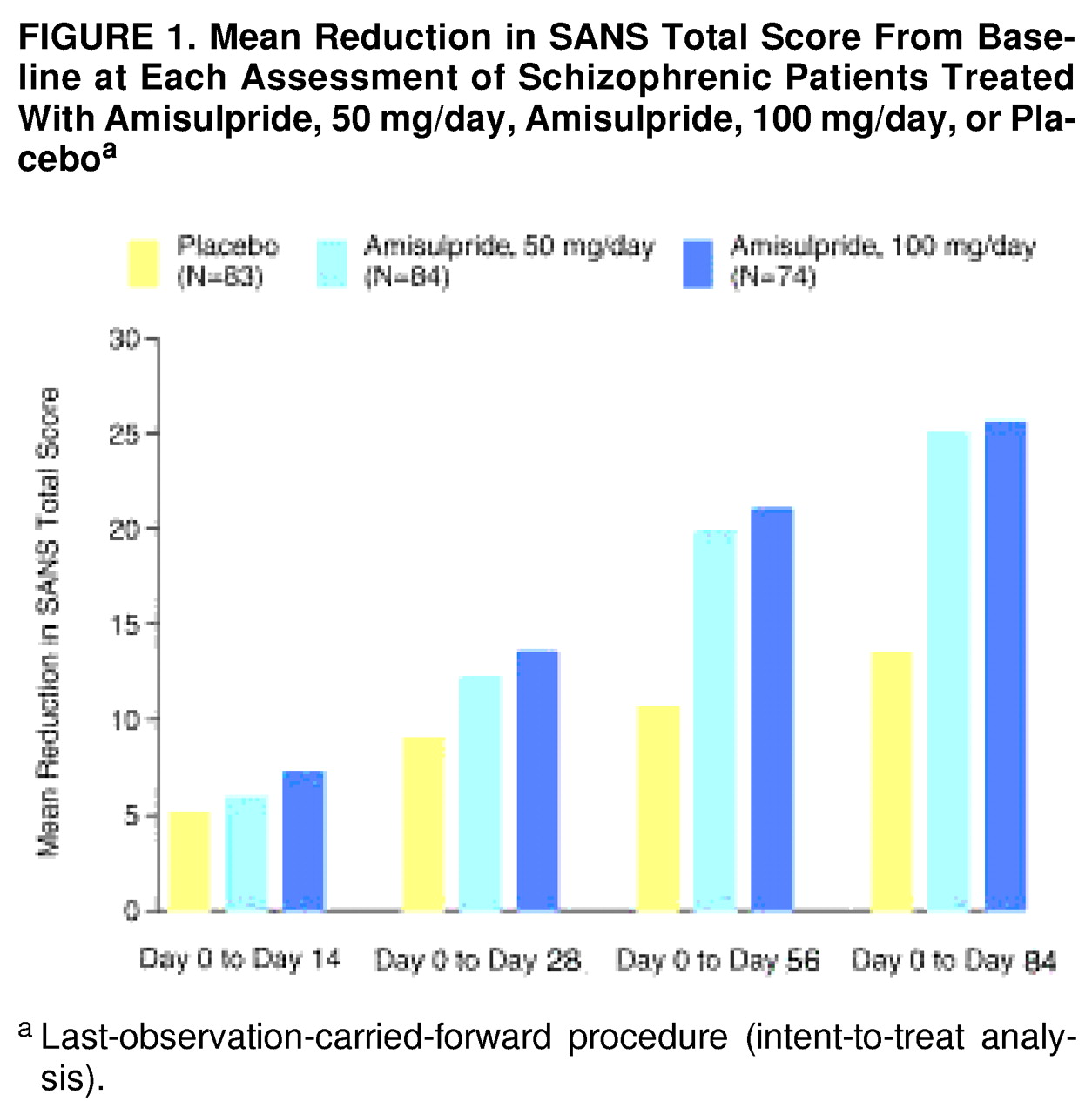
Both amisulpride-50 mg and amisulpride-100 mg patients showed a significantly greater improvement in mean total SANS score from baseline to endpoint than the placebo group (
table 2). The improvement in SANS total score at each visit is illustrated in
figure 1. Although the global difference between mean total SANS scores for the active treatment and placebo groups was statistically significant, there was no statistically significant difference between the two amisulpride groups. All five SANS component subscores (affective flattening or blunting, alogia, avolition-apathy, anhedonia-asociality, and attention) showed significant improvement in the amisulpride groups relative to the placebo group, including the factors with low scores at baseline.
Despite the modest improvements in some measures that were low at baseline, the global differences between the active treatment and placebo groups were statistically significant for secondary efficacy criteria, including scores on the SAPS, the BPRS, and the Montgomery-�sberg Depression Rating Scale. Both amisulpride groups consistently showed statistically significant differences relative to the placebo group for these secondary efficacy (
table 2), and there were no significant differences between the two active treatment groups. The difference in SAPS results was mainly due to an increase in mean scores in the placebo group.
Amisulpride-50 mg and amisulpride-100 mg patients showed significantly better results than the patients treated with placebo in terms of treatment response, as defined by “very much or much improved” on the CGI scale (item 2) at endpoint (χ
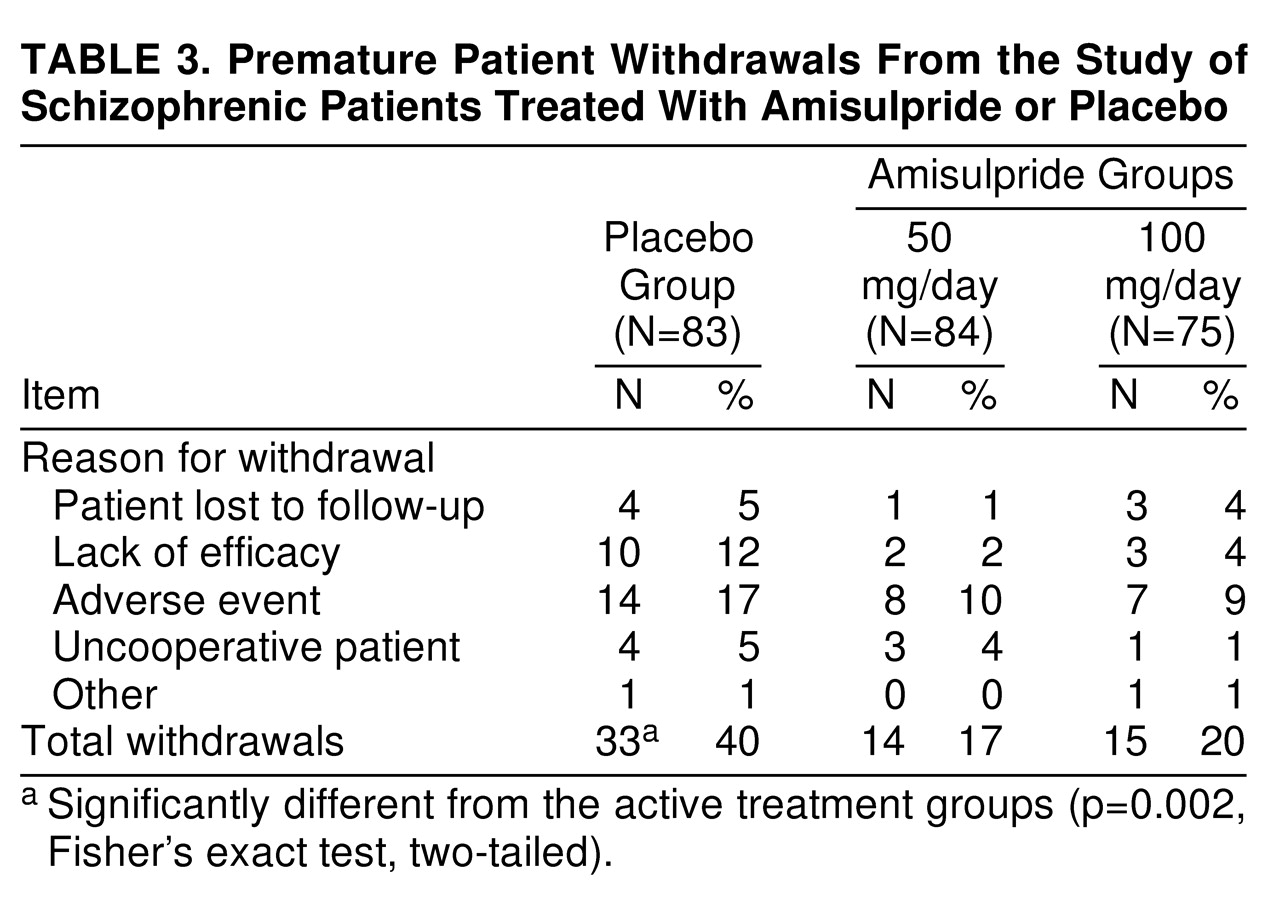
2=20.37, df=2, p<0.0001). Thus, 17 (20%) of the patients in the placebo group and 41 (49%) and 39 (52%) of the patients in the amisulpride-50 mg and amisulpride-100 mg groups, respectively, were classified as responders according to the CGI. In addition, the number of patients who withdrew from the study because of lack of efficacy was higher in the placebo group than in the two amisulpride groups (
table 3).
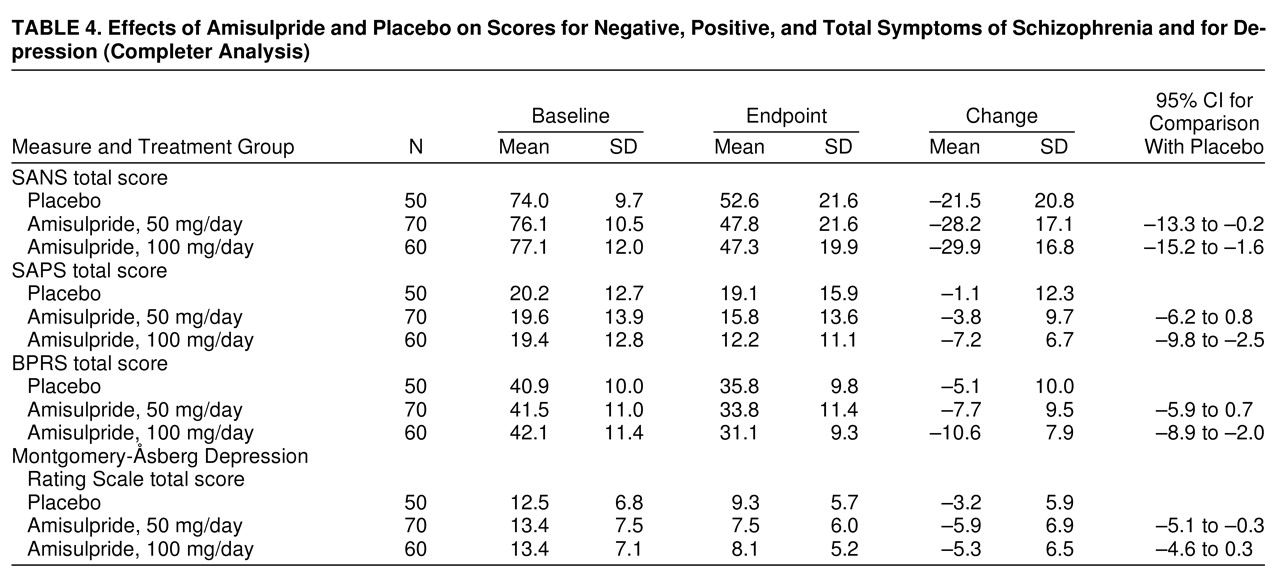
An analysis of the data on the patients who completed the study confirmed the results of the intent-to-treat analyses (
table 4).
Safety
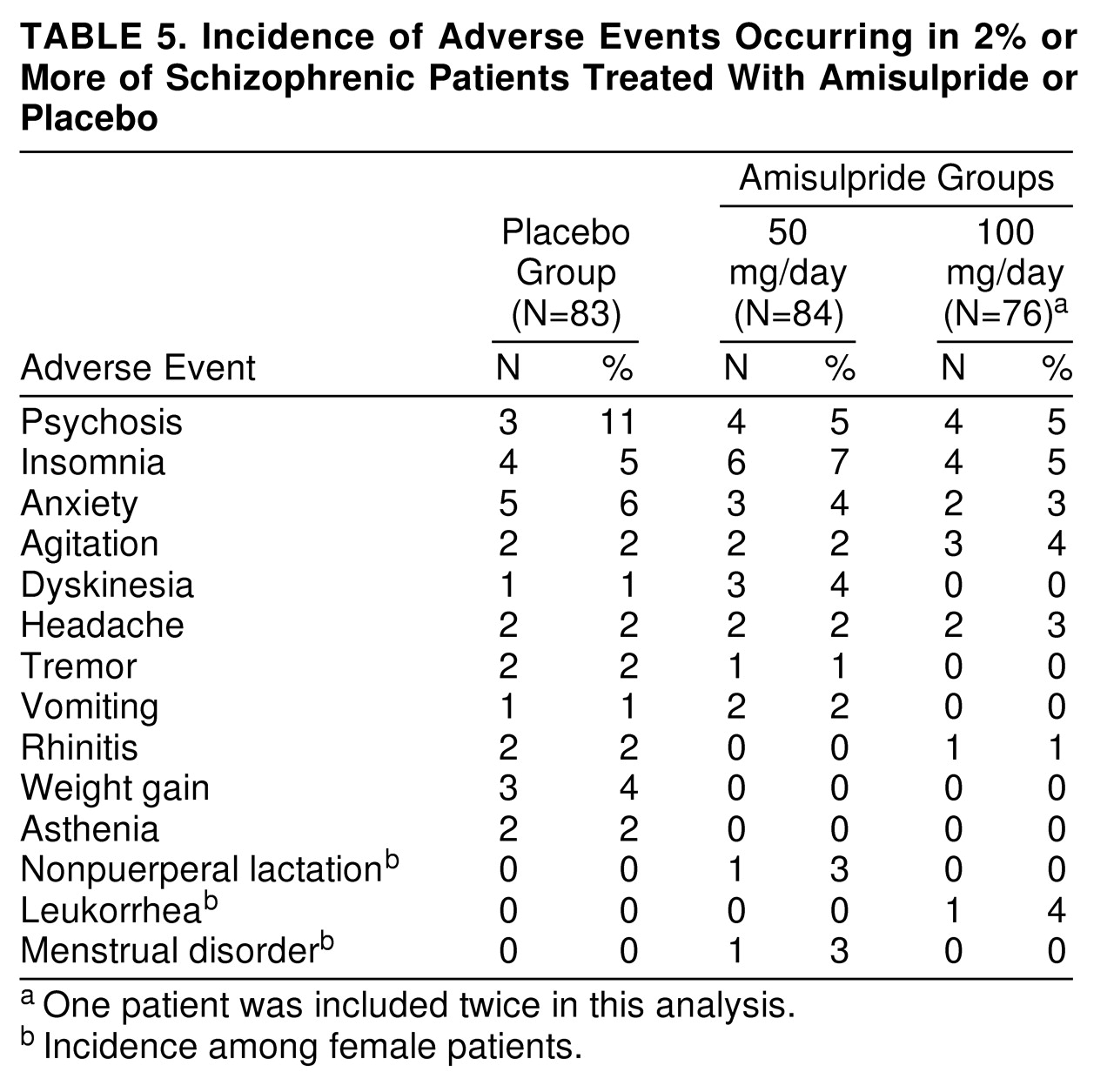
Overall, the numbers of patients with at least one adverse event during treatment were similar in all three study groups (N=27 [33%] in the placebo group, N=21 [25%] in the amisulpride-50 mg group, and N=18 [24%] in the amisulpride-100 mg group). Psychosis (reemergence of positive symptoms), insomnia, and anxiety were the most frequently reported adverse events in each treatment group (
table 5). Eight (5%) of the 160 amisulpride patients and two (2%) of the 83 placebo patients experienced at least one extrapyramidal symptom. The number of patients experiencing at least one endocrine symptom was low (N=2 [1%] of 160 in the amisulpride treatment groups).
The rate of premature withdrawal from the study because of adverse events was higher in the placebo group than in the amisulpride groups (nonsignificant difference). The main reasons for premature withdrawal in the placebo, amisulpride-50 mg, and amisulpride-100 mg groups, respectively, were psychosis (N=8, N=6, and N=3), anxiety (N=0, N=0, and N=2), and agitation (N=2, N=1, and N=1).
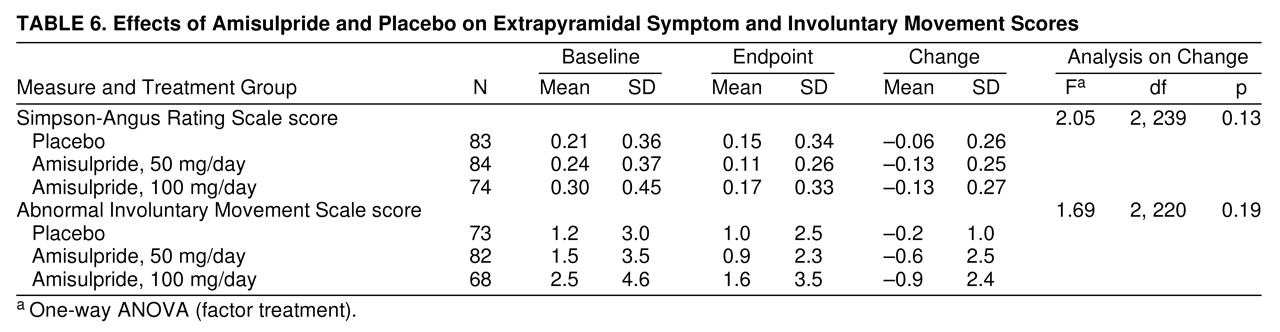
Extrapyramidal symptoms were mild at baseline (the range of Simpson-Angus Rating Scale mean item scores was 0.0–1.5) and remained mild at endpoint; no statistically significant difference in these scores between the placebo and active treatment groups was noted (
table 6). The findings were similar for the AIMS scores (
table 6). No report of akathisia occurred during the study. Only a few patients received antiparkinsonian drugs to reduce extrapyramidal symptoms: two in the placebo group (treatments were initiated 24 and 50 days after inclusion in the controlled phase of the study) and two in the amisulpride-50 mg group (in one patient, treatment was initiated during the placebo washout phase; in the other, after 45 days of amisulpride therapy). There were no significant differences between the three treatment groups in the incidence of tachycardia, bradycardia, hypertension, or hypotension during the study. With respect to body weight, 10%, 6%, and 15% of the patients in the placebo, the amisulpride-50 mg, and the amisulpride-100 mg groups, respectively, showed an increase of 5% or more at endpoint relative to baseline. Routine laboratory tests performed at baseline and at endpoint revealed no clinically significant differences between the three study groups. Finally, 90 patients of the two amisulpride groups had an amisulpride plasma assay under active treatment, yielding a mean concentration of 13.8 mg/ml (SD=2.6) for the 50-mg group and 36.5 mg/ml (SD=4.8) for the 100-mg group.
DISCUSSION
In this study, amisulpride, 50 and 100 mg/day, consistently showed a clear superiority over placebo in improving primary negative symptoms in patients with residual-type schizophrenia. The findings were similar in the two active treatment groups. Placebo control subjects were necessary because there is no recognized standard treatment for the negative symptoms of schizophrenia
(31,
32). Since baseline scores on both the SAPS and the Montgomery-�sberg Depression Rating Scale were low, and the changes in these scores were minimal, the observed differences in SANS scores cannot be explained by improvement in positive or depressive symptoms. There is probably an overlap in measurements of depression and negative symptoms that cannot be totally avoided, but the proportion of change in negative symptoms (measured with the SANS) was substantially higher than the change in depressive symptoms assessed with the Montgomery-�sberg Depression Rating Scale. The impact of change in positive symptoms on the negative symptom variation must also be considered as limited: the difference in mean SAPS scores between the placebo group and the two active treatment groups was mainly due to an increase in the SAPS mean score of the placebo group (whereas the SANS mean score of the placebo group decreased slightly). Furthermore, assessment of extrapyramidal symptoms according to the Simpson-Angus Rating Scale and AIMS scores showed that these symptoms were mild at baseline, changed minimally over the course of the study, and did not differ significantly between the active treatment and placebo groups. These findings suggest that the improvement in negative symptoms in treated patients was not the result of changes in extrapyramidal symptoms.
In view of the difficulty in defining patient populations with predominantly primary negative symptoms, it is essential to ensure that such patients have only low-grade positive symptoms and a low prominence of parkinsonian and depressive symptoms
(31). The patients evaluated in this study had met strict inclusion criteria to ensure these conditions, including a SANS score of at least 60 and a SAPS score of 50 or less. In addition, a 4-week washout period was used to eliminate the possible influence of treatment with previous antipsychotic or antidepressive agents; antiparkinsonian drugs were also discontinued during the washout period.
Amisulpride was associated with an incidence of adverse events similar to that associated with placebo, and most of these were of mild or moderate intensity. The slightly higher incidence of psychosis and anxiety in the placebo group suggests that low doses of amisulpride not only improve negative symptoms but also offer protection against exacerbation of positive symptoms. The incidence of extrapyramidal symptoms was low with amisulpride and similar to that with placebo. No clinically relevant differences in laboratory variables were observed between amisulpride and placebo patients.
The clinical improvement observed with amisulpride treatment in this study is consistent with the results of three recent placebo-controlled studies of low-dose amisulpride (mostly 100–300 mg/day) in schizophrenic patients with predominantly negative symptoms
(19–
21). In these studies, negative symptoms improved to a substantially greater extent with amisulpride than with placebo. Furthermore, in a previous 6-month study
(19), the majority of patients improved within the initial 3 months of treatment, and this improvement was subsequently maintained. As in the present study, the appropriateness of patient selection was confirmed by the low mean SAPS scores at baseline, and the improvement in negative symptoms was not accompanied by a substantial concomitant improvement in positive symptoms. In addition, parkinsonian symptom scores in these studies were low at baseline and were only minimally improved at treatment endpoint, again indicating that the improvement in negative symptoms was not secondary to changes in extrapyramidal symptoms. It is interesting that no further clinical improvement was obtained when the dose of amisulpride was increased from 100 to 300 mg/day. As noted in the present study, amisulpride was well tolerated and extrapyramidal symptoms were infrequent.
In conclusion, amisulpride proved to be superior to placebo in improving primary negative symptoms in patients with residual-type schizophrenia. Similar responses were observed with the 50-mg and the 100-mg daily doses of amisulpride. With appropriate patient selection and assessment of potentially confounding variables, we were able to distinguish the drug’s effect on primary negative symptoms from its effect on secondary symptoms. The results of this study confirm and extend the findings of previous studies with low-dose amisulpride in the treatment of patients with predominantly negative symptoms of schizophrenia. The absence of a recognized standard therapy for patients with negative symptoms of schizophrenia lends added importance to these clinical findings.
Acknowledgments
The Amisulpride Study Group includes the following participants.
In France: M.J. Amédro (Balzac); G. Amphoux (N�mes); T. Bougerol (Marseilles); M. Bourg (Bégard); B. Boussat (Bayonne); G. Clerc (Pontorson); M. Daurignac (Montauban); P. Dumont and B. Fengler (St. André-Lille); J.P. Gognau (Bailleul); P. Houillon (Plouguernevel); J.P. Kahn (Toul); P. Leclerq (Mulhouse); J.P. May (Hoerdt); R. Pagot (St. Ave); J.C. Pascal (Puteaux); C. Peretti and P. Singer (Strasbourg); P. Wuthrich (N�mes).
In Spain: A. Adam Donat (Valencia); J. Aguirre Oar (Vitoria); E. Baca Baldomero, M. Desviat Mu�oz, and T. Palomo Alvarez (Madrid); A. Bulbena Vilarrasa, G. Garcia Pares, and J.M. Costa Molinari (Barcelona); M.D. Peris Diaz (Guadalajara); M. Roca Bennasar (Palma de Mallorca); J.A. Soriano Pacheco (Málaga).
In Tunisia: S. Douki (La Manouba).
In Italy: B. Brancasi (Bari); F. Chimenz (Messina); P. Tosca (Pavia); A. Vita (Milano).