Until the early part of the twentieth century, physicians were capable of prescribing little more than placebos. It has been estimated that of the 2,000 “drugs” used in ancient China, virtually all lacked specific disease-relevant effects
(1). Cinchona bark (quinine) and foxglove (digitalis) were used in the eighteenth century, but their appropriate widespread use did not occur for centuries
(2). Although a part of medicine since its infancy, only recently have placebos played a salient role in drug development.
This article will examine the role of placebos in evaluating the efficacy of drugs used in psychiatry. I will also address the identification of placebo effects on drugs, the relevance of active placebo, the need for placebo groups in psychotherapy studies, and ethical issues concerning the use of placebo. Whenever possible, I will extrapolate what has been learned about placebos to clinical practice. My focus will be on studies of depression and anxiety disorders, but the principles addressed apply to many psychiatric disorders.
In the 1950s, the first efficacious drugs were introduced in psychiatry. In the ensuing 40 years, techniques have been developed to assess the efficacy of potentially useful compounds. Thousands of drugs have been tested, but only a hundred or so are approved for use with psychiatric disorders
(3). Most investigators suspect that useful drugs go unapproved for lack of sufficient supporting data. The unpredictable course of psychiatric disorders and our nosology of limited reliability and validity are major hurdles in establishing drug efficacy. The development of DSM-III, an atheoretical and phenomenologically based nosological system, in 1980 was an important milestone for psychiatry. However, diagnoses not based on disease pathophysiology must be inexact and include phenocopies with different prognoses. The fact that the placebo response rate varies from 25% to 60% for groups of patients with major depressive disorder is an expression of this problem
(4–
7). Furthermore, even if the pathophysiologic basis of a disorder is understood, host (i.e., individual resistance) and other poorly defined factors typically make the course of an illness variable. HIV infection is a case in point
(8). Before the development of effective treatments, some infected individuals survived 15 or more years; others had a rapid demise. Analogous unidentified prognostic factors contribute to the variable prognosis of psychiatric disorders. Thus, the difficulties in assessing a treatment’s utility in other areas of medicine are compounded by the limitations of our phenomenologically based nosological system and the lack of knowledge regarding the pathophysiology of psychiatric disorders.
How should we interpret evidence about a new treatment’s usefulness? It is worth recalling that as recently as 40 years ago, drug approval required only evidence of safety. Professional status and elaborate theorizing were sufficient to convince psychiatrists of a treatment’s usefulness
(9). Drug approval now requires an empirical, objective database.
To examine the process of drug development, it is helpful to be oriented to its three stages: in the first, safety is studied primarily; in the second, a clearer understanding of dose and utility is examined; in the third, anticipated pivotal studies establish efficacy. Knowledge gained after drug approval and wider general use is considered part of phase four.
How are data provided to satisfy these requirements, and how should the clinician interpret reports about new treatments? Briefly, phase one studies concern toxicology and establish drug safety and dose. (These data, reviewed by the Food and Drug Administration, are rarely published.) This discussion will focus on phases two and three, where drug efficacy for a specified disorder is established. There are four general classes of studies: open; drug-drug contrasts (where one drug has an established efficacy); placebo-drug contrasts, including three-armed studies (drug-drug-placebo contrasts); and discontinuation studies. In the last, a difference in relapse rates of a group of responders randomly assigned to continue drug or placebo supports drug efficacy. In an open study, an experimental drug is given to 20 to 30 patients with an anticipated response rate matching the rate of response to usual treatment; a hypothetical contrast of the proportion of responders to the new and standard drugs is possible. There are two schools of thought about open studies; one suggests they are useless because subsequent placebo-controlled studies are mandatory. However, they are easy to do and early in phase two offer a preliminary suggestion about efficacy. Open studies tend to overestimate utility, if they are not strongly suggestive of efficacy; further study is generally not warranted. Note that this applies to a “sample with an anticipated response rate”; a refractory or unrepresentative sample could result in the shelving of a useful drug.
WHY ARE PLACEBOS NECESSARY?
Discussions of types of studies set the stage for understanding the role of placebo. Once beyond early phase two studies, a placebo and treatment randomization are necessary. The ubiquitous occurrence of unidentified prognostic factors categorically eliminates patient matching as an option and makes randomization mandatory in order to prevent an imbalance between groups of unknown favorable prognostic factors. For most psychiatric disorders, no treatment can be considered effective without a placebo control because placebo response rates vary widely across patient groups. For example, the placebo response rate may be as high as 65% in people with major depression
(4–
7). Even in chronic schizophrenia, it may vary from 20% to 50% at one site, and improvement with placebo may exceed improvement with the drug at another
(10). Treatment randomization addresses unknown prognostic factors but cannot compensate for variable placebo responsiveness.
If a new drug were equivalent to the standard treatment, wouldn’t this establish its utility? Without calibrating a study group’s placebo response rate, new and old drugs might appear equally effective when, in fact, neither was having any effect. For example, a study compared amitriptyline, 150 mg/day, and tryptophan, 8 g/day, in groups of 20 patients each
(11). It was concluded that the experimental treatment was effective because tryptophan’s response rate (40%) was essentially equivalent to a standard treatment rate (amitriptyline, 55%). This study had several design errors, but inferring tryptophan’s efficacy on the basis of the amitriptyline response was probably false, since other studies failed to demonstrate tryptophan’s usefulness
(12). In this study, we suspect a placebo group would have had a similar improvement rate to that of the group receiving active treatments, preventing a false conclusion. A high placebo response rate can have an opposite, obfuscating effect, as illustrated by a study conducted in a general practice population of “depressed” patients
(13). It was concluded that imipramine was ineffective because improvement on a regimen of the drug did not exceed the placebo effect. However, the placebo response rate in the patient group studied was 59%, and the correct conclusion should have been that most of these patients did not require medication. No judgment about imipramine’s use was warranted. A placebo group is needed to accurately assess a new drug’s value.
Differences in drug-placebo improvement rates are a necessary condition to establish utility. However, merely demonstrating statistical superiority to placebo may not be sufficient to convey a drug’s clinical relevance.
Occasionally, only mean differences on a dimensional measure of psychopathology between drug and placebo are reported (i.e., a severity measure like the Hamilton Depression Rating Scale
[14], which generally varies from 0 to 30). This in itself is only somewhat informative. Virtually any group of depressive subjects, even receiving placebo, demonstrate significant improvement between baseline and end-of-study ratings on most measures (referred to as a within-group difference). Therefore, in the typical study scenario, drug and placebo groups have mean Hamilton depression scale baseline scores of 20, and at study end, the difference between the drug and placebo groups’ mean scores of 10 and 15, respectively, is statistically significant. With some drugs, there is a potential 6-point contribution on the Hamilton depression scale attributable to soporific effects, which further obfuscates the interpretation of differences on the Hamilton depression scale. Change on a dimensional measure may be difficult to extrapolate to a clinical setting. Clinical relevance is clarified by a categorical clinical judgment about patients achieving a significant improvement. A useful measure is the proportion of patients attaining sufficient improvement that pharmacologic treatment would not be changed in the usual clinical situation—i.e., the patient is much improved. A difference of 30% to 40% between drug and placebo improvement rates on this measure suggests comparability with other antidepressants. A less satisfactory, widely used measure of clinical relevance is the difference in the proportion between drug and placebo achieving a 50% reduction in the Hamilton depression scale score. This is a somewhat opaque criterion, because a 50% improvement in the Hamilton depression scale score may be obtained with residual psychopathology, requiring treatment alteration (baseline Hamilton depression scale score=22; end-of-study Hamilton depression scale score=11).
Another caveat concerns the relevance of statistical differences. Reporting a highly significant probability of a difference (i.e., p value) may convey limited information to the clinician. The concept of statistical power may clarify how a difference can be highly statistically significant but not clinically relevant. The power of a study is defined as its ability to demonstrate a statistical difference
(15). Two contributors to power are effect size—the difference in the proportion improved on drug and placebo—and group size. A relatively ineffective drug could be statistically superior to placebo if a large group is studied. Consider an extreme example: with groups of 1,000, treatment differences of 5% to 10% would be statistically significant, although not necessarily clinically relevant. Furthermore, a clinical context is required to interpret the effect size of a treatment. For example, a new treatment with an effect size of 5% to 10% would not be highly relevant when studying patients with depressive disorders but would be considered a major advance for patients with Alzheimer’s disease. Another caution: no single study provides absolute proof of efficacy. Statistics convey an idea about probability. If a statistical test suggests a probability of 0.05, it indicates that there are five chances in 100 that this finding is in error and that the two treatments are equal. Establishing a treatment’s utility requires several positive studies.
PREDICTING PATIENTS WHO WILL BENEFIT FROM PLACEBO
Numerous attempts to identify a personality type, cognitive style, or education level associated with a higher chance of responding to placebo have failed
(16,
17). Chronicity has been associated with a lower placebo response rate in depressed patients, but differences between groups are small and do not permit prediction on individual bases
(18–
21). This, in part, may explain our difficulty in predicting which patients benefit from antidepressants. Two large reviews antedating the introduction of selective serotonin reuptake inhibitors and other second-generation drugs were unable to identify consistent predictors of antidepressant response
(22,
23). The only consistent finding was that patients in the melancholic/endogenous spectrum benefited from tricyclic antidepressants. There have been no reviews that have attempted to identify predictors for the new generation of antidepressants. Our limited ability to predict which patients require antidepressants is a corollary of our inability to identify patients likely to benefit from placebos.
IDENTIFYING PLACEBO EFFECTS IN PATIENTS
Patients following a beneficial acute response, assigned to antidepressant continuation therapy for approximately 6 months, have a more favorable prognosis than those switched to placebo (or stopping treatment)
(24–
27). However, some patients desire to stop drug therapy for a variety of reasons, including increased side effect susceptibility, a desire to become pregnant, or a coexisting medical condition potentially complicating antidepressant therapy
(28–
30). Can we identify patients for whom the indication for maintenance medication may be less compelling? No predictors of survival in maintenance treatment have been clearly demonstrated. However, for patients receiving antidepressants, it may be possible to identify a greater likelihood of improvement resulting from placebo effects rather than true drug effects. The difference between placebo and true drug effects while on a drug regimen becomes apparent if we consider that 60% of a drug-treated group are rated much improved, and generally, 30% receiving placebo have equivalent improvement. Does the “much improved” drug group of 60% include the 30% who are placebo responders? This question is more complex than is initially apparent because it is unclear if drug and placebo responses are exclusive or independent. If independent, both could occur in the same patient—early improvement attributable to placebo and later a neurophysiologic “true drug” effect that sustains the euthymic state. Even if drug and placebo effects are independent, some improvement in the drug group may be attributable to placebo effects.
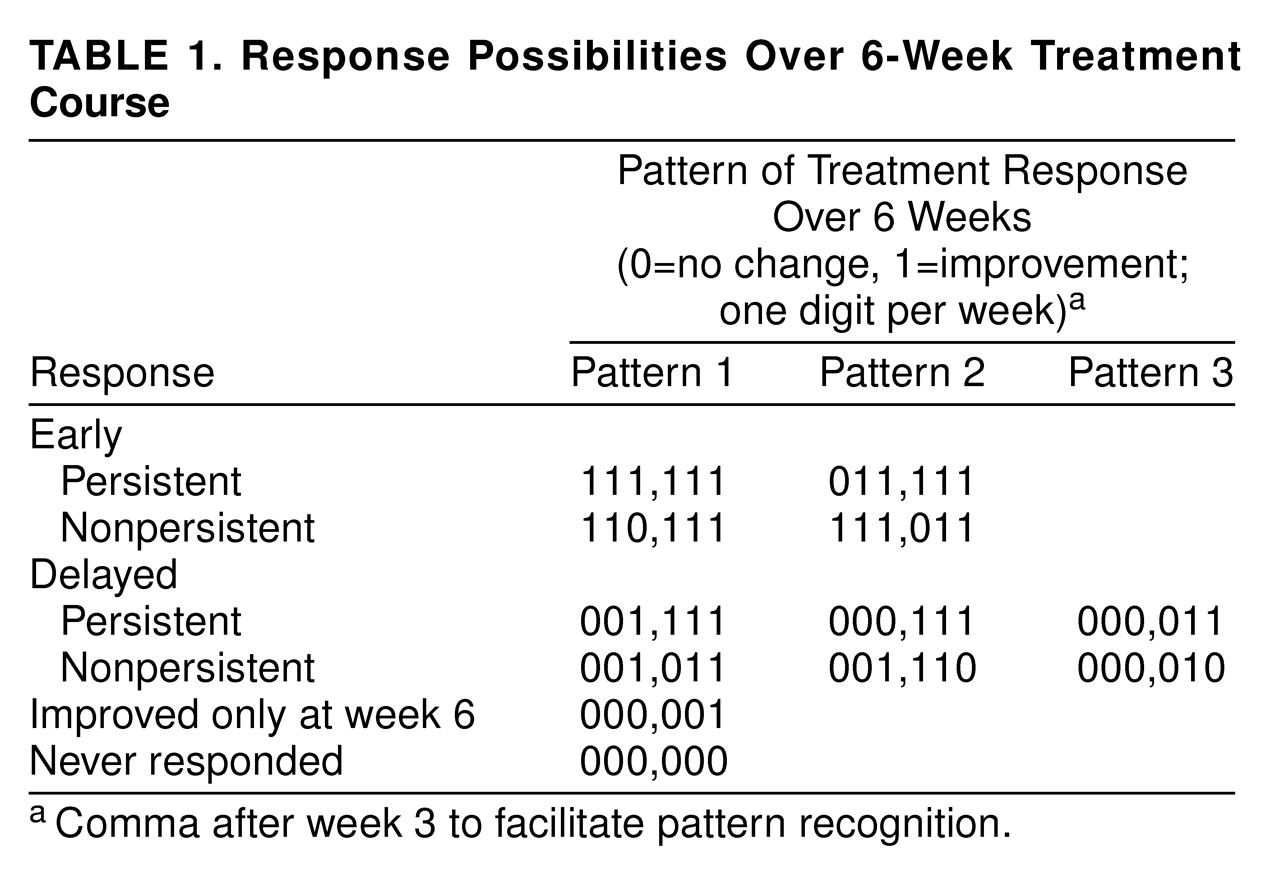
In attempting to identify true drug effects, we assumed that drug-mediated improvement would have a delayed onset and would be relatively impervious to fluctuating, unpredictable levels of patient-perceived stress and, therefore, stable. Drug responders should not have alternating good and bad weeks. A global judgment about mood improvement was made for each study week—patients much improved or very much improved were considered “improved” that week; all others were considered “unimproved.” For each patient, each week was given a 1 if the patient was improved or a 0 if the patient was unimproved. A pattern representing the 6-week treatment course was made for each patient. This pattern—111,111—characterizes a 6-week course with onset of improvement in week 1 and maintenance of improvement for all 6 weeks. The comma after week 3 facilitates pattern recognition. We refer to this as pattern analysis. In a 6-week study, there are 64 possible patterns varying from 000,000 to 111,111. In one group of approximately 200 patients, the only pattern that differed for patients taking drug and placebo was characterized by onset of improvement in weeks 3, 4, and 5 with a nonfluctuating course (see
Table 1)
(31). Thus, out of 64 possible patterns, only three distinguished between drug and placebo effects. This was repeated in a second independent patient group, and similar results have been reported by others
(32,
33). It appears relevant that three of 64 patterns repeatedly distinguish drug- from placebo-treated patients. It cannot be assumed that patients with specific patterns must have improved because of a true drug effect, only that specific patterns are associated with a greater chance of improvement resulting from drug effects than placebo effects. In the absence of understanding how drugs work, and precisely measuring this effect, pattern analysis is an aid in culling those whose improvements are more likely to result from drug than placebo effects. The converse is also true—placebo effects may cause a higher proportion of placebo patterns, but some may be the result of drug effects.
The validity of pattern analysis gains support from a study in which responders to fluoxetine were selected at random to continue on drug treatment or placebo. Patients with nonspecific patterns had identical prognoses if selected at random for drug or placebo treatment, suggesting that the fluoxetine regimen was irrelevant to their improvement
(34). Patients with a specific drug pattern had a greater likelihood of maintaining improvement if assigned to the drug compared to placebo, implicating the relevance of the drug in the genesis of their improvement. When only those continuing to take fluoxetine are considered, patients with a specific pattern compared to those with a placebo or a nonspecific pattern had a higher survival rate. This observation requires replication, however. If there is sufficient clinical reason, the clinician might be more inclined to consider discontinuing medication for a patient whose improvement was not characterized by specific patterns. This is a complicated clinical decision requiring consideration of multiple issues, including the relevant patient needs already discussed.
The implications of pattern analysis may also be useful in the clinical approach to patients relapsing during continuation therapy. This may be a particularly demoralizing experience for a compliant patient. A recent article in the lay press discussed “Prozac poop-out”
(35). Obviously, multiple effects contribute to relapse in continuation therapy, but if we consider that as much as half of the improvement observed while taking a drug may result from relatively nonpersistent placebo effects, relapse may not be because of loss of drug effect. Discussing the placebo effect may be an aid in encouraging a patient demoralized by symptom exacerbation to try an alternative drug, rather than concluding that altering drug treatment is futile.
CAN WE IDENTIFY PATIENTS FOR WHOM IMMEDIATE MEDICATION PRESCRIPTION MAY NOT BE NECESSARY?
A common design feature is a 7- to 10-day, single-blind, initial placebo run-in period before a double-blind phase. The purpose of the initial placebo run-in period is to remove patients with a high chance of a placebo effect. Typically, much improved patients do not enter a double-blind phase, although minimally improved patients may. We found that patients who minimally improved during a single-blind placebo period had a better prognosis, whether subsequently assigned to drug or placebo
(36). This was an unexpected post hoc finding and, therefore, should be viewed cautiously. However, in this group of 593 patients, there was a consistent trend across a range of diagnoses, severity, and chronicity for patients minimally improved after the initial placebo run-in period to have a more favorable prognosis. The prognosis for minimally improved patients was almost 20% better than for those who were unimproved after the initial placebo run-in period, whether the patient received drug or placebo. The relevance of this difference is put in perspective if we consider that the effect size of antidepressants is on the order of 30% to 40%.
Table 2 characterizes the treatment course of patients minimally improved after an initial placebo run-in period and subsequently randomly selected for 6 weeks of placebo. In order to estimate the morbidity experienced by these 49 patients, their 6 study weeks were stratified by the number of weeks their symptoms remained “minimally improved” (or “much improved”) versus “returned to baseline severity.” Approximately half of these patients were at least minimally improved during most of the study, having 0 or 1 of 6 study weeks in which symptoms returned to baseline severity. At study end, 79% (N=19 of 24) were deemed responders. Minimally improved patients randomly selected for placebo who had 2 or more unimproved weeks had only an 8% (N=2 of 25) chance of being rated responders at study end. A contrast of those with 1 or fewer versus 2 or more weeks (out of 6 weeks) with symptoms at baseline severity suggests that the former have a much better chance of being deemed responders at week 6 (χ
2=25.32, df=1, p<0.001). These data suggest that the patients who minimally improved after initial treatment should only receive the drug if their symptoms return to baseline severity for more than 1 week.
A total of 26% (N=154 of 593) of the patients were minimally improved after the initial placebo run-in period, and 49% (N=24 of 49) of those taking placebo had a relatively asymptomatic double-blind phase; this represents approximately 13% of the patient group. Since the number of patients studied is large, the confidence intervals are narrow, suggesting that this course characterizes a minimum of 10% of the patients.
Is this result applicable to a clinical setting, where placebo prescription is not possible? On occasion, some patients note that the onset of improvement coincides with the decision to seek treatment, and others note improvement after speaking to a caregiver for the first time. In the study described, 50% of the minimally improved group had little risk of returning to their previous level of psychopathology. A reasonable strategy in a clinical setting might be to follow patients demonstrating early minimal improvement until they have 2 unimproved weeks, since 2 symptomatic weeks separated those with good and poor prognoses. This clinical decision should take into account a variety of issues, including severity of past episodes, suicide risk, and relative contraindication to drug prescription. Although the risks associated with newer antidepressants are small, side effects do occur. It could be argued that this improvement is dependent on a pill placebo; however, some studies suggest that the mere attention of a caregiver accomplishes the same effect as a pill placebo
(25,
37). Whether this strategy would reduce morbidity and cost for some patients is questionable. Obviously, studies with prolonged follow-up would help clarify this question. In addition, this post hoc observation would have greater validity if replicated.
CONTRASTING MEDICATION AND PSYCHOTHERAPY: THE ROLE OF PLACEBOS
It has been suggested that some types of psychotherapy are effective in depressive and anxiety disorders
(38). In studying the relative merits of drug and psychological approaches, a frequently used study design consists of groups receiving psychotherapy, medication, and a combination of the two. The virtually ubiquitous result is that all groups have an equal outcome, although occasionally a superior outcome has been reported for a psychological approach
(39–
41). The adequacy of the psychopharmacologic treatment in these studies has been questioned
(42). A more basic debate involves the type of conclusions that are possible when studying drugs and psychotherapy in the absence of a placebo group
(43–
46). Klein has made a convincing argument that a placebo group is necessary to internally calibrate a group’s responsivity to nonspecific treatment effects
(46). The appropriateness of medication prescription for any study group is oblique unless the placebo response rate for that group is known. Since the utility of antidepressants has been repeatedly demonstrated, unless the group studied already demonstrates a drug-placebo difference, conclusions about the relative efficacy of psychotherapy in a drug-responsive study group are not warranted. Consider the study conducted in general practice (referred to previously), with high response rates for subjects taking placebo and imipramine. Had that group been used to study psychotherapy versus imipramine, it might have been concluded that the treatments were equivalent, rather than the correct conclusion—that neither treatment was having a specific effect. We suspect that some transient dysphoria might demonstrate a higher response rate to placebo than drug. Clearly, some patients benefit from psychotherapy and do not require drug treatment. It is also obvious that all schools have their therapeutic allegiance, and regardless of one’s intentions, bias may alter patient evaluations. How, then, to study the relative merits of different treatments? The suggestion has been made that studies contrasting psychotherapy, medication, and placebo be done at multiple sites having known therapeutic allegiances. Results that replicate across sites should be considered valid.
ARE ACTIVE PLACEBOS NECESSARY?
Several recent reviews suggest that an active—i.e., symptomatic—placebo is necessary to estimate antidepressant efficacy
(47,
48). This position was originally proposed by Thomson
(49), on the basis of a review of double-blind studies of tricyclics using active and inactive placebos. Thomson concluded that in 14% (N=1 of 7) of studies using an active placebo, but in 59% (N=40 of 68) of those using an inert placebo, the medication was found to be effective. The obvious implication is that in the absence of an active placebo (usually atropine), the estimate of antidepressant efficacy is inflated. Four other studies with an active placebo in addition to the six originally reviewed by Thomson were found
(50–
59). The rationale for giving an active placebo would be to augment the effect observed with an inactive placebo, making it more difficult to attribute drug benefit to nonspecific, rather than specific, drug effects. Therefore, if side effects are relevant in enhancing therapeutic effects, a higher response rate would be anticipated with an active placebo. This is not the case. When the data were pooled from those studies in which a judgment could be made about the proportion of responders, it was found that 22% (N=60 of 276) of patients taking active placebos were judged responders
(50–
59). One study
(55) with a placebo response rate of six out of 90 appears to be an outlier. The remaining studies have a response rate of 30% (N=55 of 186). This is virtually identical to the response rate seen with inactive placebos. The weak effect size observed in some of the studies using active placebos appears to result from a less-than-expected proportion of responders in the drug group. This is because of these studies’ design flaws. Specifically, eight of the 10 studies, completed when drug trial design was in its infancy, are flawed by the design shortcomings of this period (early 1960s). Virtually all of these trials violate at least one basic psychopharmacologic tenet: antidepressant dose is critical, and a 4-week antidepressant trial duration underestimates drug efficacy. Studies demonstrating that 300 mg of imipramine or its equivalent is superior to 150 mg of imipramine within a patient group, as well as others that demonstrate equal import of dose effects for monoamine oxidase inhibitors, established the importance of adequate dose
(60–
63). Furthermore, two studies reported that between 4 and 6 weeks of fixed dose treatment, there was a statistically significant increase in drug benefit versus placebo effects
(64,
65). Of the 10 studies using active placebo, the study duration was less than or equal to 4 weeks for five studies
(50,
51,
53,
56,
58), and for two others
(54,
57) the drug dose was less than or equal to 150 mg of imipramine or its equivalent. In addition, Hussain
(52) did not discuss dose or duration. It is reasonable to conclude that available evidence does not provide a compelling case for the necessity of an active placebo. Research on the effects of an active placebo would have to compare outcome with active and inactive placebos to clarify the merits of each.
IS PLACEBO USE ETHICAL WHEN EFFECTIVE TREATMENTS EXIST?
The Declaration of Helsinki, although somewhat vague, appears to contradict the use of placebos if an effective treatment is known
(66). Other authorities clearly stated that if an effective treatment is known, a placebo group is contraindicated
(67). Some even suggested that editors refuse to review papers in which a placebo is used for a disorder for which there is an effective treatment
(68). I think that placebo use is acceptable in disorders characterized by a fluctuating course with only a slight chance that a delay in effective treatment would result in permanent damage and where patients are closely monitored.
Most psychiatric disorders have a fluctuating course. There is no evidence that treatment delay results in permanent damage to the patient (analogous to an arthritic joint freezing). The public health implications of approving an ineffective treatment are of overwhelming importance. Certainly, that will delay effective treatment for many who are not being closely monitored and are, therefore, at greater risk. If efficacy equivalent to a standard treatment is considered definitive, ineffective drugs could be approved
(69–
71). An example from an article by Leber
(69) characterizes four studies with a standard drug (desipramine or imipramine), a new drug, and placebo. The new drug has been approved abroad. In these four trials, if no placebo group were included, the experimental drug would appear identical to the standard. The placebo group suggests that these trials failed to establish the efficacy of the experimental drug. This type of result is referred to as a “failed study,” meaning that a drug with established efficacy is not found to be superior to placebo, as opposed to a negative study, in which a new drug is found ineffective but a standard drug is found to be superior to placebo.
Once a drug is approved, it is possible that hundreds of thousands of patients would receive an ineffective drug; a gross estimate of potential mortality associated with this is possible. Consider that as many as 15% of depressed patients die as a result of suicide. If the period of risk is 30 years, it is possible that as many as 0.5% of patients per year will take their lives. It is reasonable to conclude that some avoidable suicides would be attributable to use of an ineffective drug. It is harder to estimate the morbidity associated with an approved ineffective treatment. Testing a new drug requires approximately 500 patients receiving placebo for 6 weeks while being closely monitored by experts. However, approval of an ineffective drug could expose hundreds of thousands of patients to ineffective treatment with ordinary clinical review. An ineffective drug has obvious public health consequences. We are ethically bound to minimize patient risk in research of this type. Studies in which patients may receive placebos should be carefully monitored; if there is evidence of significant deterioration, patients should be removed from the study. Patients should only be treated with placebo for some finite period of time and should receive the best possible care for the remainder of the episode. Best possible care should be exhaustive and include systematic use of all relevant treatments until an effective one is found. It is hoped that this approach will reduce the morbidity and mortality of all patients with these diseases.
SUMMARY AND CONCLUSIONS
Understanding the neurophysiological basis of placebo would be an important clue about change in both normal and pathological mood. There is some evidence that endorphins may be relevant, but a full discussion of that mechanism is beyond the scope of this article, and the interested reader is referred to an article by Benedetti and Amanzio
(72). It is clear that psychiatric disorders have a fluctuating course, that a phenomenologically based nosological system is inexact, and that the interaction between the two leads to a large proportion of patients in any group experiencing a placebo effect.
It may also be possible to identify patients whose improvement is a result of a placebo effect. This is at best an educated and inexact guess; however, under certain circumstances, this may be an aid in planning the treatment of patients who have a contraindication to continuing a psychopharmacological regimen.