In general, evidence-based psychotherapies, specifically cognitive behavior therapy (CBT) and interpersonal psychotherapy, demonstrate comparable efficacy to pharmacotherapy in treating mild to moderate depression
(1 –
3), although the evidence supporting the use of these psychotherapies in populations with severe depression is less established
(4 –
6) . Venlafaxine differs from selective serotonin reuptake inhibitor (SSRI) antidepressants by virtue of its ability to inhibit both serotonin and norepinephrine reuptake transporters
(7,
8) . Compared with SSRIs, venlafaxine has either comparable or superior rates of remission in the treatment of major depressive disorder
(9 –
11) .
Functional neuroimaging techniques, including fluorine-18-fluorodeoxyglucose positron emission tomography (PET) have helped to delineate regional differences in metabolic activity between depressed and nondepressed subjects
(12,
13), as well as between depressed and remitted states
(14 –
16) . In the case of antidepressant medications, most investigators have focused on within-subject changes in cerebral metabolism before initiating and during treatment with various SSRIs or dual action antidepressants
(15,
17 –
19,
20 –
22) . Increased dorsolateral and decreased ventrolateral prefrontal metabolism have been reported most frequently in antidepressant respondents
(15,
20 –
24) .
There have been fewer investigations of changes in cerebral activity following psychological interventions
(25 –
28) . Limited evidence suggests predominantly right-sided decreases in prefrontal activity, with conflicting reports on anterior cingulate direction of change
(25,
29) . Nevertheless, these investigations provide support for the psychobiological impact of both psychotherapy and pharmacotherapy. Issues of nonrandomization and differences in response rates between medication and psychotherapy groups as well as differences in the duration of respective treatments have confounded previous reports
(29) .
In order to address these limitations, we carried out a randomized controlled trial to compare the patterns of change in cerebral metabolism in depressed patients following treatment for 16 weeks with CBT or venlafaxine. The specific aims of this study were twofold: 1) to delineate change patterns that distinguish responders to CBT from responders to venlafaxine and 2) to identify common regional metabolic changes associated with the response status with either treatment modality.
Based on our previous findings, we hypothesized that response to either treatment modality would be associated with changes in metabolism previously identified as differentiating the depressed and remitted states; response to CBT would be associated with additional prefrontal decreases reflecting the primacy of altered cognition
(27,
29) ; and, in contrast, response to venlafaxine would be associated with subcortical activation, corresponding to neurovegetative symptom reduction
(26,
30,
31) .
Method
Participants
Thirty-one patients (13 men and 18 women) were recruited at the Centre for Addiction and Mental Health, a fully affiliated teaching hospital at the University of Toronto, Ontario, Canada. They were required to be between 20 and 50 years old; meet DSM-IV criteria for major depressive disorder; be currently in a major depressive episode as assessed by the Structured Clinical Interview for DSM-IV, Patient Edition (SCID-I/P)
(32) ; and score 20 or greater on the 17-item Hamilton Depression Rating Scale (HAM-D)
(33) . They were also required to be free of any antidepressant medication for at least the preceding 2 weeks (4 weeks for fluoxetine) and be in good physical health with no evidence of neurological or other unstable medical conditions. Other axis I diagnoses, including concurrent anxiety disorders and substance abuse or dependence within the past 6 months, evidence of active suicidal ideation, pregnancy, and previous failure to respond to an adequate trial of CBT or venlafaxine were exclusion criteria. All subjects provided written informed consent for participation in this study, which was approved by the Research Ethics Board of Centre for Addiction and Mental Health.
Treatment
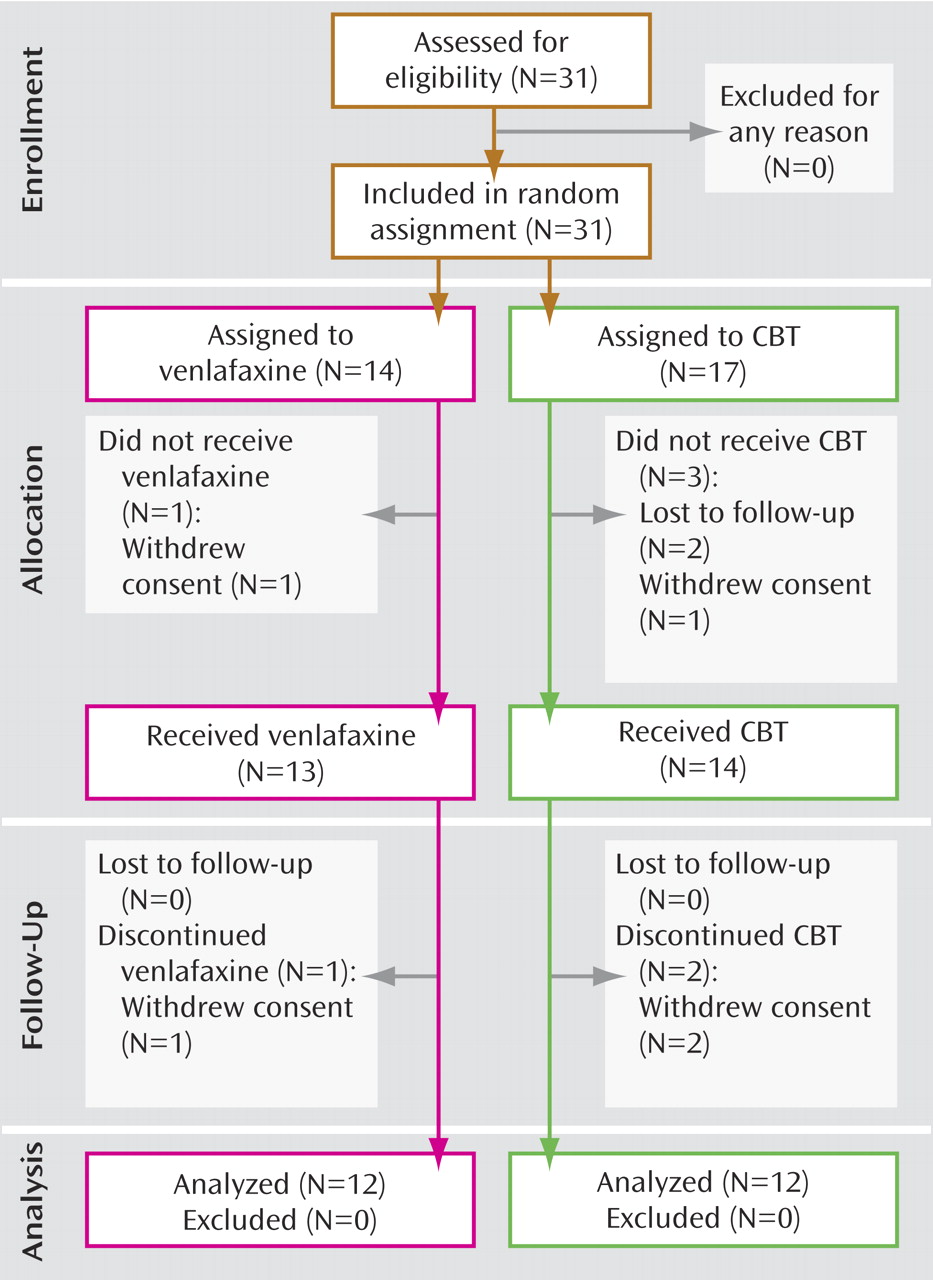
Study participants were randomly assigned to receive either venlafaxine 75 mg/day to 225 mg/day (N=14) or CBT (N=17) for up to 16 weeks. Two subjects from the venlafaxine group and five subjects from the CBT group failed to complete a minimum of 8 weeks of treatment and a second PET scan (
Figure 1 ). Subjects in the venlafaxine group received 75 mg daily for the first 2 weeks, thereafter increasing to a target dose range between 150 mg and 225 mg (
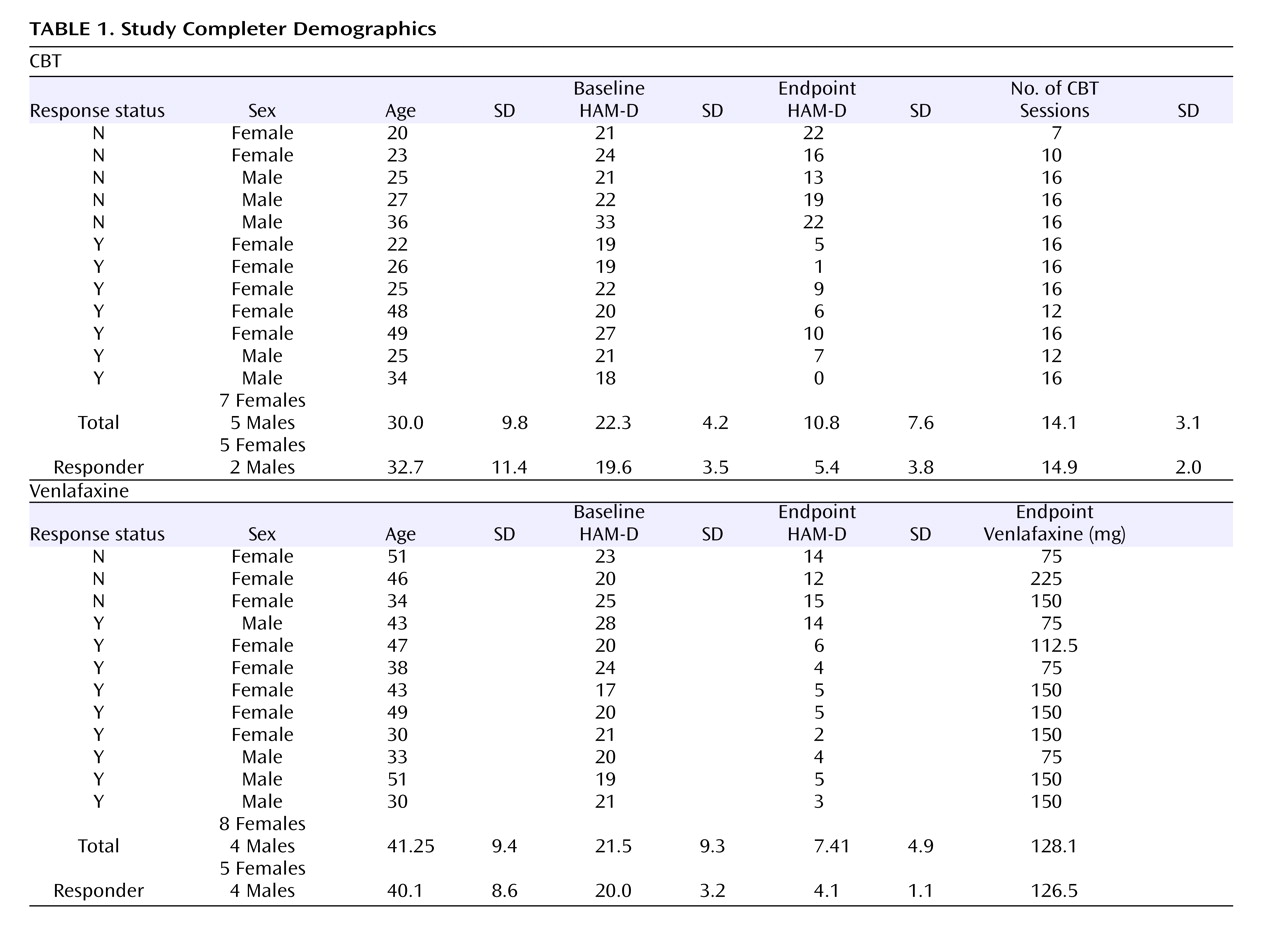
Table 1 ).
Subjects in the CBT group received individualized outpatient sessions on a weekly basis, administered by a trained CBT therapist (Dr. Lau or Dr. Bieling, who have 12 and 10 years of experience, respectively, in delivering manual-based therapy according to the manual of Beck et al.
[34] ). All CBT sessions were audiotaped to enable ratings of treatment fidelity, which was confirmed by the supervising psychologist (Dr. Segal). Subjects undergoing CBT were taught to use a number of therapeutic strategies intended to reduce automatic reactivity to negative thoughts or attitudes and to combat dysphoric mood.
Therapists used a collaborative inquiry technique to guide subjects to a more evidence-based and less reactive reconstruction of their experience.
All subjects were evaluated biweekly using the HAM-D. Response was defined as a minimum reduction of 50% in HAM-D scores from baseline, and remission was defined as a HAM-D endpoint score <7.
PET Procedure
PET measurements of regional cerebral glucose metabolism were obtained at baseline and again at the end of treatment, using standard imaging methods that have been previously described
(29) . Scans were acquired during the week before treatment initiation and within 1 week of the last treatment visit. For each scan, a 5 millicurie (mCi) (185 megabecquerels [Mbq]) fluorodeoxyglucose dose was injected intravenously, with image acquisition beginning after 40 minutes (PC 2048b; GEMS-Scanditronix, Uppsala, Sweden). All scans were acquired at a consistent time between 9 a.m. and noon, with subjects in a supine, awake, and resting state, with their eyes closed and ears uncovered. Subjects were asked to refrain from food, coffee, or alcohol intake for a minimum of 8 hours before each scan session.
Participants were given no explicit cognitive instructions but were asked to avoid ruminating on any one topic during the fluorodeoxyglucose uptake period. Wakefulness was additionally monitored every 10 minutes by a study investigator. Emission data were acquired during a 35-minute period (approximately 1 million counts per slice; 10 cm field of view). A customized, thermoplastic face mask was used to minimize head movement for the initial scan and for accurate repositioning at the second session. Raw images (15 parallel slices; 6.5 mm center-to-center interslice distance) were corrected for attenuation, reconstructed and smoothed to a final in-plane resolution of 7.0 mm at full width at half-maximum.
Data Analysis
Statistical analyses were performed using statistical parametric mapping (SPM) (SPM99-Wellcome Department of Cognitive Neurology, London) implemented in Matlab (version 7.0; Mathworks Inc., Sherborn, Mass.)
(35) . The data were first screened for distributional properties, outliers, and missing values. All scans were normalized to the Montreal Neurological Institute International Consortium for Brain Mapping 152 stereotactic template within SPM99, which references brain locations in three-dimensional space relative to the anterior commissure. The images were then corrected for differences in the whole-brain global mean and smoothed using a Gaussian kernel to a final in-plane resolution of 12 mm at full width at half-maximum. Absolute glucose metabolic rates were not calculated.
Since response-specific CBT or venlafaxine effects were the primary foci of this study, results in this report are first presented for those participants (venlafaxine: N=9; CBT: N=7) who met the a priori-defined response criteria, and, subsequently, for those participants who did not meet a priori-defined response criteria (venlafaxine: N=3; CBT: N=5). To identify regional metabolic changes unique to each treatment modality, the contrast of prepost contrasts between CBT and venlafaxine treatments were evaluated to identify clusters meeting the p<0.01 height threshold for seven a priori-defined regions (orbitofrontal cortex [Brodmann’s areas 11, 47], dorsolateral prefrontal cortex [Brodmann’s area 9], anterior and posterior cingulate cortices, thalamus, striatum, and amygdala) and p<0.001 uncorrected for all other brain regions that also exceeded the minimum expected cluster size in SPM. Conjunction analyses
(36) were also employed to identify the colocalization of significant baseline-endpoint changes that were common to both CBT- and venlafaxine-treated subjects, separately for responders and nonresponders.
The resulting t values were converted to z scores, with brain locations reported as x, y, and z coordinates in Montreal Neurological Institute space with approximate Brodmann’s areas identified by mathematical transformation of SPM99 coordinates into Talairach space.
Results
Clinical Effects
Twelve participants in each group completed at least 8 weeks of treatment and received both baseline and endpoint PET scans (
Figure 1 ). In the CBT group, the mean HAM-D scores were 20.6 (SD=3.4) at baseline and 9.8 (SD=7.6) at endpoint, with a mean drop in severity of 53% (t=4.83, df=11, p=0.001). In the venlafaxine-treated group, the mean HAM-D scores were 20.3 (SD=3.0) at baseline and 7.4 (SD=4.9) at endpoint, representing a mean symptom reduction of 64% (t=8.13, df=11, p<0.001). There were no statistically significant between-group differences at either baseline (p=0.851) or endpoint (p=0.193). Nine of the 12 venlafaxine-treated subjects met criteria for response, and eight met criteria for remission. In the CBT group, seven of 12 subjects were classified as responders; of these, five met criteria for remission (
Table 1 ). With the exception of one CBT responder, who received 12 weeks of treatment, all responders received 16 weeks of treatment.
Neuroimaging Effects
Responders
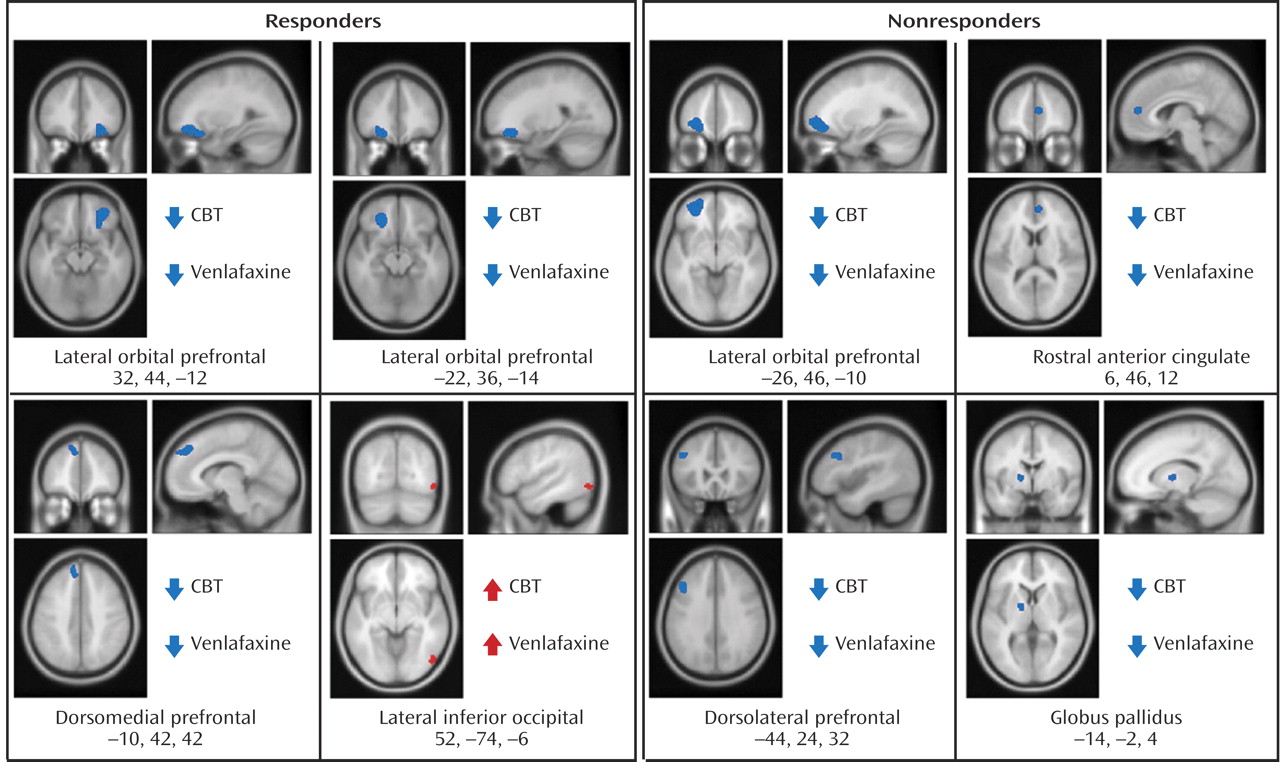
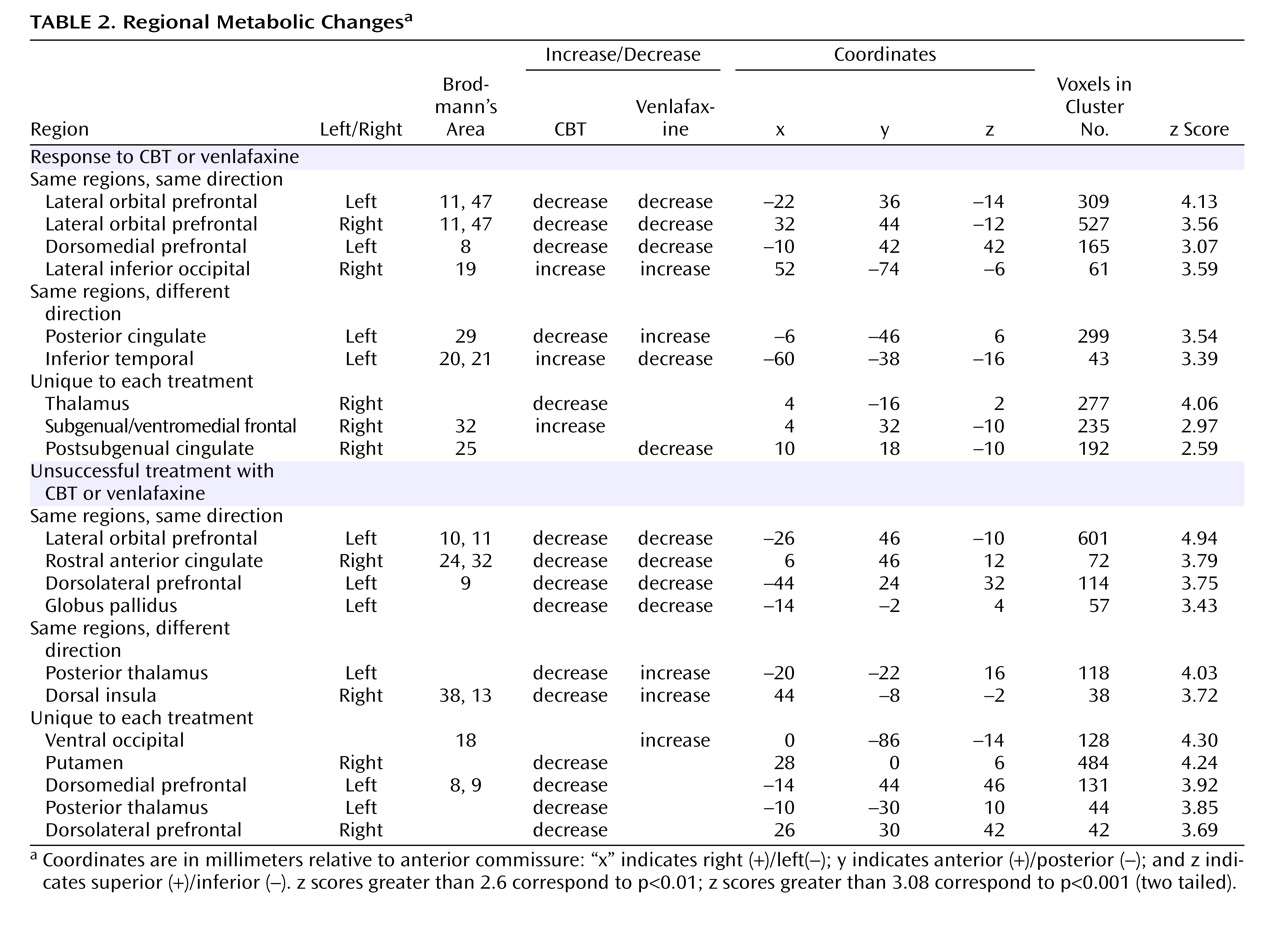
Before treatment initiation, there were no statistically significant between-group differences in brain glucose metabolism between subjects assigned to either CBT (N=12) or venlafaxine (N=12) (data not shown). Conjunction analysis identified the following changes common to both CBT and venlafaxine responders: decreased metabolism in the left and right lateral orbital frontal cortex (Brodmann’s areas 11, 47), the left dorsomedial prefrontal cortex (Brodmann’s area 8), along with increased metabolism in the right inferior occipital cortex (
Figure 2,
Table 2 ).
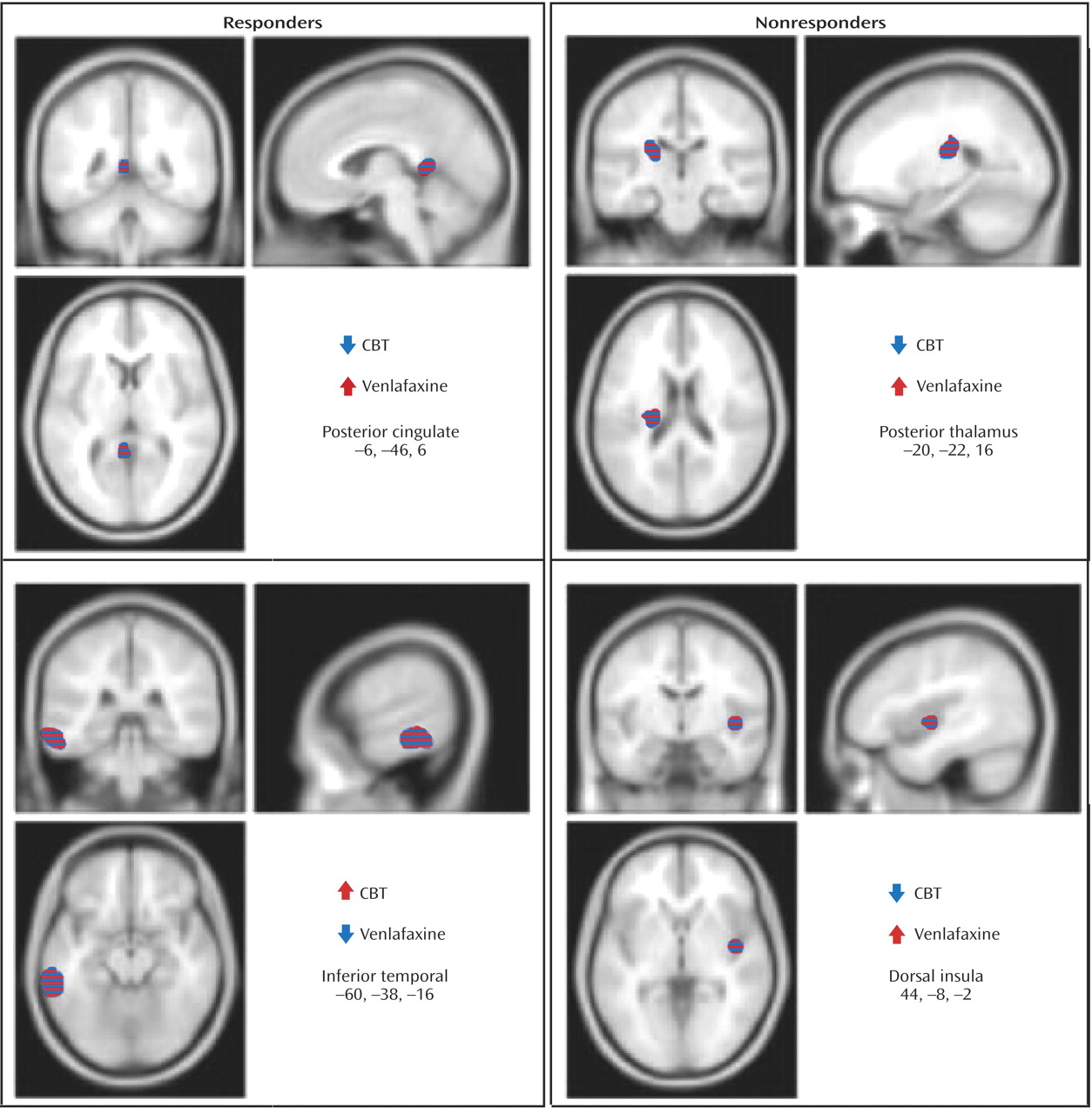
Response to treatment was also associated with differential changes in metabolism in several other regions. Whereas response to venlafaxine was associated with increases in the posterior cingulate (Brodmann’s area 29), CBT responders displayed decreased metabolism. Conversely, differential metabolic changes were also found in the left inferior temporal cortex (Brodmann’s areas 20, 21), increased with CBT and decreased with venlafaxine therapy (
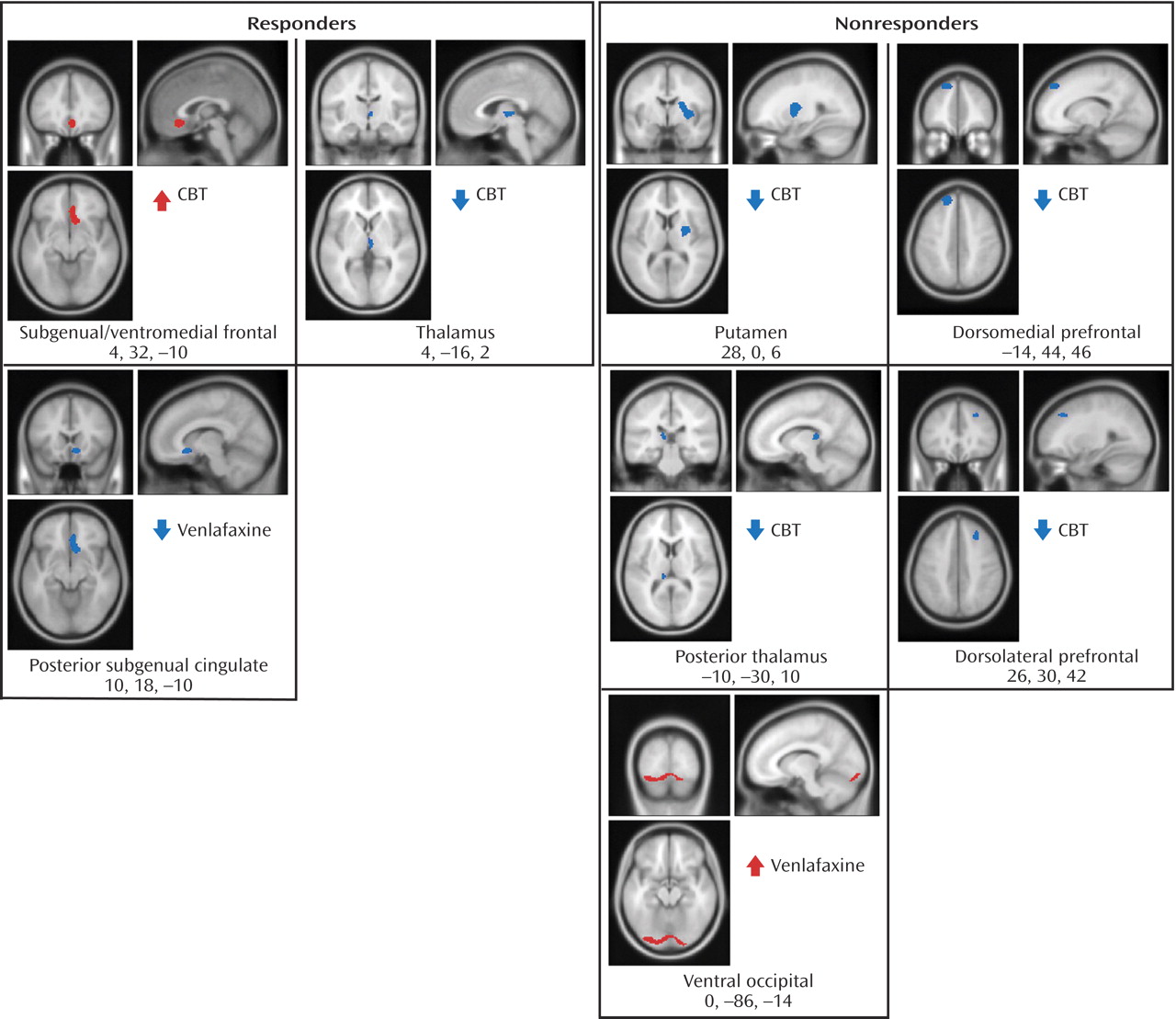
Figure 3 ). Additionally, metabolic changes unique to each treatment modality were observed. Decreased metabolism in the thalamus was found to be exclusive to CBT responders, while decreases in a region encompassing the right nucleus accumbens and a posterior part of the subgenual cingulate (Brodmann’s area 25) were only observed in venlafaxine responders. Responders to CBT displayed unique increases in metabolism in a region encompassing a more anterior portion of the subgenual cingulate/ventromedial frontal cortex (Brodmann’s area 32), as well as the right occipital-temporal cortex (Brodmann’s area 19), that were not observed in venlafaxine responders (
Figure 4 ).
Neuroimaging Effects
Nonresponders
Left lateral orbitofrontal and left dorsolateral prefrontal decreases, along with decreases in the rostral anterior cingulate and globus pallidus, were common to both CBT and venlafaxine nonresponders (
Figure 2,
Table 2 ). Differential changes in metabolism were also observed between the CBT and venlafaxine nonresponder groups. Whereas nonresponse with venlafaxine treatment was associated with increases in the posterior thalamus and the dorsal insula, CBT nonresponders displayed decreased metabolism in these same brain regions (
Figure 3 ).
Metabolic changes unique to each treatment modality were also noted within nonresponders. Decreases in the thalamus, putamen, dorsomedial and dorsolateral prefrontal cortices and the posterior thalamus were observed in the CBT group, while venlafaxine nonresponders were limited to increases in the ventral occipital cortex and dorsal cerebellum (
Figure 4 ).
Discussion
To our knowledge, this is the first report to evaluate changes in regional glucose metabolism following an extended 16-week randomized comparative trial of pharmacotherapy and CBT. These results extend our previous findings comparing CBT with paroxetine
(29) . In the present study, however, participants were randomly assigned to a treatment group, both groups received 16 weeks of treatment, a duration that reflects standard CBT practices, and response rates for the antidepressant and CBT were comparable.
Common Regions
The colocalization of common regional brain metabolic changes associated with response to either CBT or venlafaxine represents treatment-independent effects of clinical response. Response to either treatment was associated with decreased metabolism in the ventrolateral and dorsomedial prefrontal cortices, changes that have previously differentiated depressed and euthymic states in major depressive disorder populations
(16,
19,
25,
29) . Differences in metabolic brain activity between baseline and endpoint in nonresponder groups included a combination of unique and similar metabolic brain changes (albeit of reduced magnitude) to those observed in responders. Preclinical data implicate the ventrolateral and orbitofrontal cortices in the modulation of behavioral and visceral responses to defensive, fearful, and reward-directed behaviors
(37,
38) . Through anatomical projections to neurons in the amygdala, striatum, hypothalamus, and other limbic and brainstem structures, the orbitofrontal cortex modulates responses to aversive or appetitive stimuli and integrates experiential stimuli with emotional salience
(39,
40) . Moreover, emotional processing biases in depressed patients have also been shown to map onto the orbitofrontal cortex
(41,
42) . In our analysis, response to either treatment was associated with a reduction in metabolic activity bilaterally in the orbitofrontal cortex, while decreased metabolism restricted to the left orbitofrontal cortex was observed irrespective of treatment group or treatment outcome.
The dorsomedial prefrontal cortex has been implicated in the self-referential processing of emotional stimuli. The right dorsomedial prefrontal cortex is activated in a wide range of emotional tasks, including recollection of affective-laden personal life events
(43), attention to subjective feeling
(44), and processing of emotion-related meanings
(45) . All of these tasks share an explicit representation of different aspects of the self and integration of these aspects with emotional reactions and experience
(46) . Decreases in dorsomedial prefrontal cortex activity, reported exclusively in the CBT group in our original study
(29), were observed in both groups in this current investigation, perhaps reflecting the additional noradrenergic modulation by venlafaxine or the longer study duration. Nonresponse to either treatment was also associated with metabolic decreases in the dorsal prefrontal cortex, but in a region lateral and inferior (Brodmann’s area 9) to the locus identified in responders (Brodmann’s area 8). Moreover, nonresponse to either treatment was associated with decreased metabolism in the rostral anterior cingulate (Brodmann’s areas 24, 32) and left globus pallidus. Reduced activity in the rostral anterior cingulate has been previously reported as a predictor of treatment nonresponse
(47 –
49) .
CBT Findings
Previously, we reported that response to CBT was associated with increased activity in the hippocampus and dorsal anterior cingulate (Brodmann’s area 24c) as well as decreases in the dorsolateral (Brodmann’s areas 9, 46), ventrolateral (Brodmann’s areas 47, 11), and medial (Brodmann’s areas 9, 10, 11) frontal cortices
(29) . In this new cohort, increases were localized to the anterior subgenual/ventromedial frontal cortex anterior cingulate (Brodmann’s area 32), while decreases were observed in comparable prefrontal regions (ventrolateral [Brodmann’s areas 47, 11] and dorsomedial [Brodmann’s area 8] frontal cortices). An accumulating evidentiary base supports disruptions in limbic–thalamic–cortical circuits, and limbic–cortical–striatal–pallidal–thalamic circuits as principal mediators of cognitive impairments in mood disorders
(19,
48,
50) . In contrast to our previous report, in this study we also observed decreases in the right thalamus accompanying CBT response, whereas metabolic decreases were localized to the left thalamus in CBT treatment nonresponders. Additional metabolic decreases in CBT nonresponders included bilateral reductions in the dorsolateral prefrontal cortex and the right putamen and globus pallidus. Analysis of the baseline-endpoint changes in subjects who did not respond to CBT may delineate metabolic patterns characterizing the effects of CBT administration, rather than antidepressant response to CBT treatment.
Venlafaxine Findings
Decreases in glucose metabolism in the ventromedial frontal cortex
(17,
20) and increases in the temporal cortex
(18,
25) have been previously reported in SSRI responders. Venlafaxine-specific effects found in the subgenual cingulate (Brodmann’s area 25), ventrolateral prefrontal and temporal cortices, posterior cingulate (Brodmann’s area 29), and putamen replicate findings from studies of other antidepressant medications
(22,
26,
29,
31) .
Venlafaxine is a dual-action antidepressant that blocks the reuptake transport of both serotonin and norepinephrine and, as such, could be expected to demonstrate more extensive cortical engagement than previously observed single-action antidepressants. Serotonergic neurons in the dorsal raphe nucleus give rise to long axons that project throughout the brain
(51), while the locus coeruleus in the pons is home to the majority of noradrenergic neurons with axonal projections to prefrontal cortical and subcortical sites, including the putamen, thalamus, and hippocampus
(52) . Variations in these regions across studies likely reflect study design differences in dose, treatment duration, and response variability
(53) . Our report of increased metabolism in the posterior thalamus, dorsal insula, and occipital cortex in subjects not responding to venlafaxine may reflect a specific SNRI-treatment effect.
Psychotherapy-Pharmacotherapy Differences
In the previously reported post hoc comparison of brain changes associated with response to either CBT or paroxetine
(29), there were the following baseline-endpoint changes within the cingulate cortex: increases in the dorsal anterior cingulate (Brodmann’s area 24c) with CBT treatment and decreases in the subgenual cingulate (Brodmann’s area 25) with response to paroxetine. These findings are replicated in the present study, with decreased right posterior subgenual cingulate activity in venlafaxine responders. Increased metabolism with CBT was localized to a different portion of the subgenual cingulate anterior, dorsal, and medial to the region identified with venlafaxine (
Figure 4 ).
Decreased metabolism in the posterior subgenual cingulate has also been associated with response to SSRIs
(19,
22,
25) and electroconvulsive therapy
(54) . Decreased subgenual cingulate activity with symptom abatement is consistent with other clinical neuroimaging data, reflecting a positive correlation between metabolic activity in the subgenual anterior cingulate and depression severity
(55,
56) . Taken together, this has provided the rationale for the subgenual cingulate (Brodmann’s area 25) as a target for deep brain stimulation in a treatment-resistant group of depressed patients
(57) .
Evidence from preclinical and human studies supports extensive reciprocal connections between the subgenual cingulate and areas implicated in the expression of behavioral, autonomic, and endocrine responses to stressors, aversive stimuli, and rewarding stimuli
(40) . Dysfunction of the ventral anterior cingulate cortex in primary mood disorders may thus contribute to the altered emotional behavior and neuroendocrine function evident in depression. Divergent subcortical findings of increased metabolism within the posterior cingulate in the venlafaxine group, compared with decreases accompanying response to CBT, may be associated with increased noradrenergic tone with venlafaxine treatment, leading to increased locus coeruleus-subcortical input
(58) . An analysis of treatment nonresponders further supports this hypothesis. Increases in the posterior thalamus and dorsal insula were reported in venlafaxine nonresponders, in contradistinction to the observed decreases associated with unsuccessful CBT treatment.
Putative Mechanism Mediating Changes in Glucose Metabolism
A possible explanation for the observed changes in glucose metabolism at the cellular level is an alteration in glutamate signaling
(59) . Astrocytes, situated proximal to both cerebral capillary beds and neuronal synapses, play a pivotal role in the coupling of glucose uptake and glutamatergic neurotransmission
(60) . Following the release of glutamate by the presynaptic neuron, its activity on the postsynaptic surface is quickly terminated by glutamate reuptake into the surrounding glia
(61) . Subsequent glutamate reuptake by glia stimulates glucose uptake and metabolism by astrocytes, adenosine triphosphate-generating processes which ultimately lead to the recycling of glutamate back to glutamine
(50) . In keeping with this view, changes in neurotransmission, represented by glutamatergic synaptic activity, are believed to reflect differences in the fluorine-18-fluorodeoxyglucose-PET signal
(62) .
Intracellular glucose delivery is dependent on several isoforms of facilitative glucose transporters
(63) . Exposure to some dual-action antidepressants has been linked to increases in glucose transporter messenger ribonucleic acid (mRNA) levels
(64), whereas exposure to some tricyclic antidepressants may actually inhibit glucose transport
(65) . Recently, greater scrutiny of the metabolic effects of psychotropic medications has highlighted the association between some noradrenergic antidepressants, hyperglycemia, and increased sensitivity to endogenous counterregulatory mediators
(66) .
Limitations
There are several limitations to this investigation. Our relatively small cohort size may have precluded the identification of subthreshold changes in brain activity because of power constraints, particularly in our analysis of nonresponder changes. We did not acquire arterial radioactivity levels, preventing a calculation of absolute glucose metabolism. In the absence of high-resolution structural magnetic resonance images, region-of-interest analyses were not performed. While the homogeneity of this population represents a strength, it may also limit generalizability of our findings to real-world populations of depressed patients where medical and psychiatric comorbidities are frequently encountered.
Another limitation is the acquisition of resting-state scans, rather than obtaining brain responses to affective or cognitive challenges before and following treatment. Recent data suggest that this latter approach can reveal residual relapse vulnerability following successful pharmacological or cognitive intervention
(67) . Since we did not acquire neuroimaging data early in the course of treatment (e.g., after 1 week), we were unable to examine the potential predictive value of early change on subsequent clinical outcome. Finally, we did not include self-rated psychometric evaluations to complement symptom measurement with the HAM-D.
Clinical experience suggests that some patients respond better to psychotherapy and others to pharmacotherapy. This study helps to build an emerging evidence base for differential changes in regional brain activity following response (and nonresponse) to pharmacotherapy compared with psychotherapy
(13,
21,
25,
28,
29) . Exploration of baseline differences between venlafaxine and CBT responders may also help to understand which patients are best served by each of the therapeutic interventions that were evaluated.