Method
Subjects
Twelve adolescents and young adults with an adolescent-onset alcohol use disorder (five male and seven female subjects; mean age=17.2 years [SD=2.2]) and 24 healthy comparison subjects (10 male and 14 female subjects; mean age=17.0 years [SD=2.4]) were recruited (age range for all subjects=13.5–21.0 years). Because of the high degree of known variability in volume of brain structures
(9,
13), each subject with an alcohol use disorder was case matched with two healthy comparison subjects for age (within 6 months; z=0.1, df=1, p=0.92) as well as for sex and handedness. Subjects with alcohol use disorders were similar to the comparison subjects with respect to height (mean=170.0 cm [SD=11.7] and 169.3 cm [SD=9.9], respectively; t=0.17, df=34, p=0.87); weight (mean=68.3 kg [SD=11.1] and 71.1 kg [SD=17.5]; t=0.50, df=34, p=0.62); and full-scale IQ (mean=109.0 [SD=10.5] and 110.5 [SD=17.6]; t=0.28, df=34, p=0.78). The socioeconomic status of the two groups also did not differ (mean=34.4 [SD=6.3] and 38.5 [SD=9.9]; t=1.29, df=34, p=0.20), with the mean scores indicating that both groups were on average from working class families. The comparison group was recruited by advertisement from the community and had no lifetime histories of psychiatric disorders, including alcohol and substance use disorders.
Substance use disorder diagnoses were determined by using a modified form of the Structured Clinical Interview for DSM-IV (see reference
14 for details). Information regarding alcohol- and drug-related areas was gathered by means of direct interviews, since family informants typically underreport alcohol and drug involvement
(15).
Other axis I mental disorders were assessed by using a modified version of the Schedule for Affective Disorders and Schizophrenia for School-Age Children (K-SADS)
(16) and the K-SADS Epidemiologic Version (K-SADS-E)
(17) interviews, with both adolescent and parent(s) as informants. An expanded assessment of posttraumatic stress disorder (PTSD) was completed as part of the K-SADS Present and Lifetime Version (K-SADS-PL)
(18).
Consensus diagnoses were reached among the interviewer, assessment coordinator, and a clinically experienced faculty psychiatrist or psychologist by using the best-estimate method
(19–
21). A date of onset, defined as the time at which diagnostic criteria were first met, was determined for each disorder
(22). The alcohol use disorders group consisted of seven subjects with lifetime alcohol dependence and five subjects with lifetime alcohol abuse. Comorbid conditions included other substance abuse or dependence (cannabis [N=9] or hallucinogens [N=2]), major depressive disorder (N=9), conduct disorder (N=5), PTSD (N=5), attention deficit hyperactivity disorder (ADHD) (N=5), oppositional defiant disorder (N=2), generalized anxiety disorder (N=2), and bipolar disorder (N=1). The mean age at onset for an alcohol use disorder was 15.9 years (SD=2.4). While the mean age at onset for the nine subjects with cannabis abuse was 15.3 years (SD=2.3), for seven of these subjects the alcohol use disorder diagnosis preceded their cannabis use disorder diagnosis. Brain structural volumes for four of the subjects with alcohol use disorders (with comorbid PTSD) and 10 of the healthy comparison subjects were previously reported
(8).
Subjects were excluded from the study if the following were found: use of drugs or alcohol during the 2 weeks before the magnetic resonance imaging (MRI) scan, assessed by a urine drug screen and alcohol breathalyzer test within 12 hours of the scan; presence of a clinically significant medical or neurological illness; gross obesity (weight greater than 150% of ideal body weight) or growth failure (height under third percentile); full-scale IQ below 80; pregnancy; or inability to consent to the protocol because of insufficient English skills. After a complete description of the study was given to the subject and parents if appropriate, written informed consent was obtained. Subjects received monetary compensation for participation.
MRI Acquisition
MRI scans were performed at the University of Pittsburgh Medical Center Magnetic Resonance Research Center by using a Signa 1.5-T system (GE Medical Systems, Milwaukee). A sagittal scout series verified subject position, cooperation, and image quality. A three-dimensional, spoiled gradient recalled acquisition in the steady state pulse sequence was used to obtain 124 contiguous images with slice thickness of 1.5 mm in the coronal plane (TE=5 msec, TR=25 msec, flip angle=40˚, acquisition matrix=256 ¥ 192, number of excitations=1, field of vision=24 cm). Coronal sections were obtained perpendicular to the anterior commissure/posterior commissure line to provide a more reproducible guide for image orientation. Axial proton density and T2-weighted images were obtained to enable exclusion of structural abnormalities on the MRI scan. A neuroradiologist reviewed all scans and ruled out clinically significant abnormalities. All subjects tolerated the procedure well. No sedation was used.
Image Analysis
The imaging data were transferred from the MRI unit to a computer workstation and analyzed using the IMAGE software (version 1.52) developed at the National Institutes of Health
(23), which provides valid and reliable volume measurements of specific structures by using a semiautomated segmentation approach. All measurements were made by trained and reliable raters who were blind to subject information. Independent designation of regions for all structures described had interrater reliabilities that ranged from 0.91 (corpus callosum regions) to 0.99 (intracranial volume) and intrarater reliabilities that ranged from 0.97 (corpus callosum regions, amygdala and hippocampal volumes) to 0.99 (intracranial volume). Complete methodological details have been previously described
(8) and are only briefly presented here.
Intracranial volumes were calculated by summing areas of successive coronal slices, including gray and white matter and CSF volumes, and multiplying by slice thickness. Cerebral volumes were measured in the same manner after exclusion of cerebellum and brainstem.
Total cerebral gray and white matter volumes were calculated by using a semiautomated segmentation algorithm. This computerized segmentation technique uses mathematically derived cutoffs for gray matter-white matter CSF partitions with histograms of signal intensities.
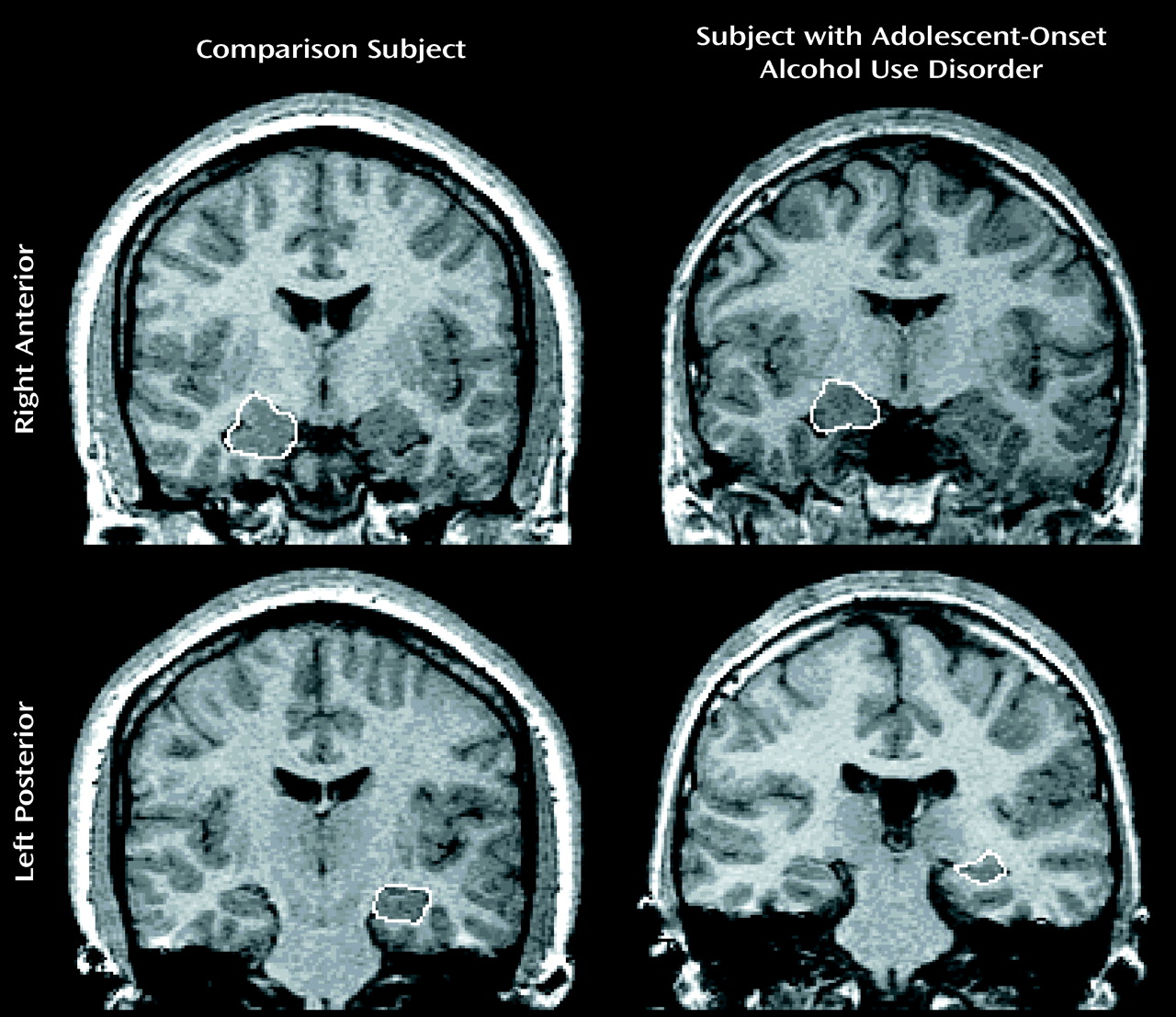
The amygdala and hippocampus were manually traced in the coronal plane. Then the areas were summed for each slice and multiplied by slice thickness, using the most anterior slice in which the temporal stem was first visible. The most anterior slice showing the mammillary bodies was used as the amygdala-hippocampal boundary. The ambient cistern defined the medial border of the anterior hippocampus. The superior border of the hippocampus was bounded anteriorly by the temporal horn and posteriorly by the fornix. Our measurement of the hippocampal formation included the cornu ammonis, dentate gyrus, and subiculum.
Right and left lateral ventricle measurements were obtained using a manual tracing technique in the coronal plane. Measures were summed for each slice and multiplied by slice thickness. This technique was used rather than measurements of CSF to reliably exclude the choroid plexus.
The corpus callosum was identified from a single midsagittal section selected as the slice showing full visualization of the anterior and posterior commissures and the cerebral aqueduct
(8). This area was divided into seven regions (rostrum, genu, rostral body, anterior and posterior midbody, isthmus, and splenium).
Statistical Methods
Data distributions were examined for normality using Shapiro and Wilks’s W statistic. Where significantly nonnormal distributions were found (amygdala and lateral ventricle volumes), the data were log transformed to normalize the distributions before applying parametric tests. In cases where no transformation normalized the data (age), nonparametric tests were used. Categorical demographic variables were compared using t tests or Fisher’s exact tests as appropriate. Between-group differences were tested by means of analysis of variance after controlling for intracranial volume. In testing for covariate effects, such as sex and interactions (sex by group), multivariate regression analysis was used. Brain structures for which adjusted least squares means differed significantly between the groups (total hippocampus) were correlated with clinical data by using Spearman correlation coefficients. All significance tests involving the main hypothesis were two-tailed, with alpha=0.05. A reduced critical value of p<0.01 was used in our exploratory analyses of sex, sex-by-group, and clinical correlation data to control for type I errors.
Discussion
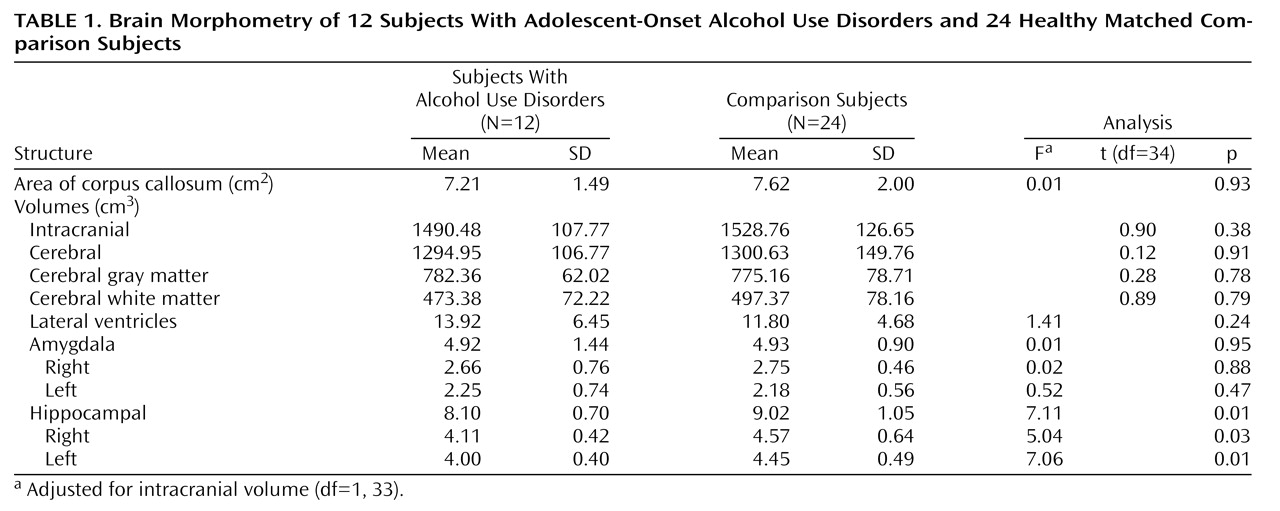
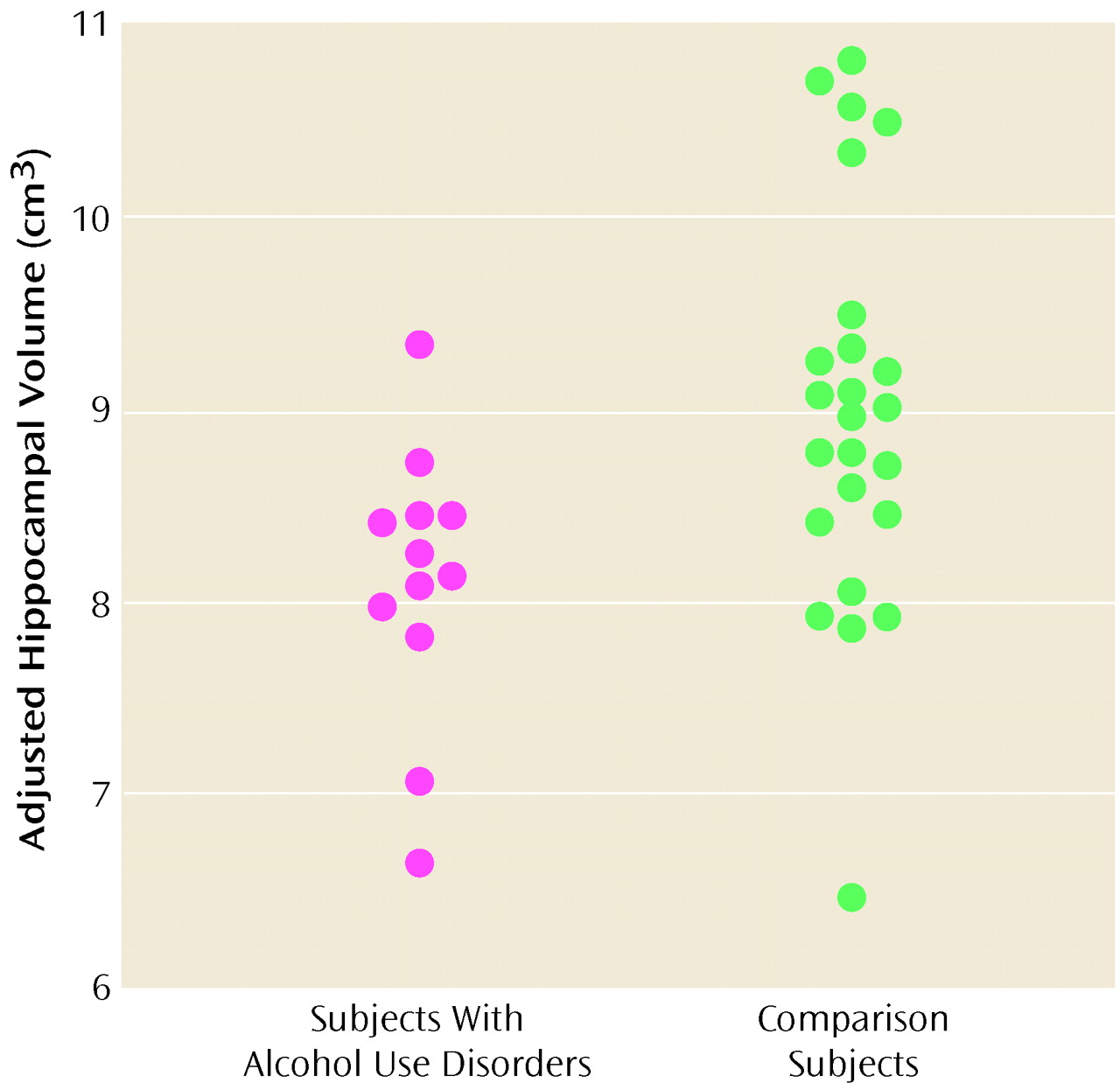
Adolescents and young adults with adolescent-onset alcohol use disorders were found to have significantly smaller left and right hippocampal volumes than healthy matched comparison subjects. Intracranial, cerebral, cortical gray and white matter volumes, right and left amygdala volumes, and total corpus callosum area and its comparison regional measures did not differ between groups. Hippocampal measures adjusted for intracranial volumes correlated positively with the age at onset and negatively with the duration in years of the alcohol use disorder. There were no significant correlations between hippocampal measures and alcohol consumption. To our knowledge, this is the first study to examine the association of an adolescent-onset alcohol use disorder with the brain morphology of adolescents and young adults.
There are several possible explanations for the finding of decreased hippocampal volumes in adolescents and young adults with adolescent-onset alcohol use disorders. Alcohol-related neurotoxic effects on the hippocampus may be mediated by N-methyl-d-aspartate (NMDA) receptors, a class of excitatory amino acid receptors, or by the neurotrophic functions of NMDA receptors, thus causing disrupted hippocampal development in adolescents. A smaller hippocampus also may indicate the presence of another risk variable common to alcohol use disorders and smaller hippocampal size (i.e., comorbidity). Finally, a preexisting smaller hippocampus may itself be a risk factor for an adolescent-onset alcohol use disorder.
Smaller hippocampal volumes in adults with alcohol use disorders than in adults without alcohol use disorders have been reported
(24–
26). In animals, hippocampal damage is seen with chronic alcohol administration
(27,
28). Alcohol may have direct toxic effects on the hippocampus through NMDA receptors that are independent of seizure activity
(29). Findings from animal studies showed that the hippocampus exhibits enhanced NMDA-mediated neurotoxicity during alcohol withdrawal
(30,
31). Chronic alcohol consumption leads to up-regulation of NMDA receptors
(32). Alcohol withdrawal leads to increased excitatory transmission. This process can result in discrete excitotoxic neuronal damage due to repetitive, excessive stimulation of NMDA receptors
(33,
34). Furthermore, chronic intermittent alcohol exposure in rats results in hippocampal cell loss, an effect not seen in animals constantly exposed to alcohol
(35,
36). Adolescents with alcohol use disorders tend to binge drink
(37). Since hippocampus maturation continues during adolescence and young adulthood, the pattern of adolescent drinking may interact with aspects of hippocampal development. Alternatively, inhibition of NMDA receptors by ethanol may result in a complex disinhibition of several excitatory pathways that lead to a neuron-necrotizing reaction of cortical and limbic structures
(38). This latter toxic mechanism is age dependent and becomes fully sensitive between puberty and adulthood
(39). Furthermore, NMDA receptors are critically involved in the process of activity-dependent synaptic elimination in the adolescent brain
(40). This plasticity declines during adolescence in parallel with reduction in NMDA receptor activity. Hence, disrupted hippocampal development may be the result of reduced NMDA receptor activity and, consequently, a disruption of its neurotrophic functions. Thus, the adolescent hippocampus may be particularly vulnerable to the effects of alcohol through NMDA-mediated mechanisms.
High comorbidity is typical with adolescent-onset alcohol use disorders
(22). As in our cohort, other studies have shown that a substantial percentage of adolescents with alcohol use disorders have other comorbid substance use disorders (particularly cannabis abuse) as well as conduct disorder, ADHD, mood disorders, and PTSD
(22). Our subjects with alcohol use disorders had high rates of comorbid major depression. Smaller hippocampal volumes were reported in clinical conditions thought to be associated with increased corticosteroid levels. A substantial proportion of adult major depressive disorder patients exhibit cortisol hypersecretion (for review see reference
41). However, cortisol hypersecretion is not strongly associated with either major depressive disorder or dysthymia in children and adolescents
(42). One study showed that depressed prepubertal children had lower cortisol secretion during the first 4 hours after sleep onset with no overall 24-hour differences between depressed subjects and a comparison group
(43). Decreased hippocampal volumes have not been consistently shown in adults with major depression
(44,
45), but a recent study found decreased hippocampal volumes in adult women with recurrent major depression
(46). These findings correlated with longer duration of depressive illness and were not associated with substance abuse. No sex-by-group effects were seen in hippocampal volumes in our subjects with alcohol use disorders.
Smaller hippocampal volumes were reported in adults with Cushing’s syndrome
(47) and conditions of traumatic stress such as combat veterans with PTSD
(48,
49), adult PTSD related to childhood abuse
(50), and female adult survivors of childhood sexual abuse
(51). Among adolescents with alcohol use disorders, PTSD is typically preceded by a history of physical or sexual abuse
(22,
52–
54). In the adult PTSD studies, the incident rates of comorbid lifetime diagnoses of alcohol abuse and dependence were high, ranging from 71% to 76%. These investigators controlled for lifetime alcohol consumption by including subjects in their comparison group with alcohol use disorders and using standardized lifetime alcohol use interviews. However, in a cross-sectional investigation, we showed by means of MRI scans that maltreated children and adolescents with PTSD (N=43) had smaller brain structural measures of intracranial volumes, midsagittal areas of the corpus callosum, and larger ventricular volumes than nonmaltreated healthy comparison subjects (N=61)
(8). The predicted decrease in hippocampal volume was not seen in these young PTSD subjects. In these adult PTSD studies and our child PTSD study
(8), maltreated subjects with PTSD exhibited high degrees of comorbidity, particularly for mood disorders. For example, 36 (83.7%) of 43 child and adolescent PTSD subjects had a depressive disorder. However, maltreated children and adolescents with PTSD did have less comorbid histories of alcohol and substance abuse, as only four of our adolescent subjects had this history
(8); they were included in this present study. In the present study, we found a suggestive although nonsignificant decrease in hippocampal volume in the alcohol use disorders subgroup without PTSD, when compared with their matched comparison subjects. Ethanol has been shown to stimulate pituitary corticotrophs resulting in hypercortisolemia, a feature of heavy drinking
(55,
56). The hippocampus is also rich in glucocorticoid receptors, and a high level of cortisol is damaging to the hippocampus
(57). Comorbid adolescent-onset alcohol use disorders may contribute to some of the differences in hippocampal findings between children and adolescents and adults with PTSD. Alternatively, an interaction effect of an adolescent-onset alcohol use disorder and PTSD may be especially toxic to the hippocampus.
In MRI studies of children and adolescents with ADHD, no hippocampal differences were seen between those with ADHD and healthy comparison subjects
(58). No hippocampal differences were seen between healthy matched comparison subjects and child and adolescent subjects who had generalized anxiety disorder without comorbid PTSD or a substance use disorder
(59). To date, there are no published MRI hippocampal studies on adolescent conduct disorder or cannabis dependence. Cannabis dependence may be associated with 9-tetrahydrocannabinol-induced cell death in preclinical studies (for review see reference
60) and is a potential confound in our study. Smaller hippocampal volumes are seen in childhood-onset schizophrenia
(61) and pediatric seizure disorders
(62,
63). However, subjects with a history of psychosis were excluded from our study. Seizures and nutritional deficiencies may account for findings of decreased white matter and hippocampal volumes in alcohol-induced brain damage in adult alcohol use disorders. However, nutritional deficiencies and alcohol withdrawal seizures are very rare in adolescents
(19) and were not seen in our subjects with alcohol use disorders.
Thus, hippocampal toxicity in adolescent-onset alcohol use disorders may be mediated by several mechanisms. Since we found that total hippocampal volume correlated positively with the age at onset and duration of an alcohol use disorder diagnosis, one may wonder if adolescence is a more vulnerable period for hippocampal toxicity. Still, the small cohort and effect sizes and extensive comorbidity in our subjects with alcohol use disorders may have contributed to these findings. Furthermore, preexisting smaller hippocampal volumes may be a risk factor for an adolescent-onset alcohol use disorder, regardless of comorbidity. Our preliminary findings suggest, but do not prove, that the adolescent hippocampus may be particularly susceptible to the effects of alcohol. However, this study did not have adequate power to examine comorbidity within subjects with alcohol use disorders, and further study of these issues is warranted.
Lezak
(64) and Welsh and Pennington
(65) have described the correlation between the hippocampus and the integrity of various memory functions that include storage and recall, rote learning, memory for verbal material with context, and spatial memory. In this study, we did not examine hippocampal function, so we do not know if our findings have clinical sequelae. Neurotoxic effects to the brain from chronic excessive drinking may be reversible
(12,
66–
68). This recovery process may be especially important for adolescent brain development, since adolescents with an alcohol use disorder who become sober may have a better chance for recovery of function than in adulthood because of the developmental plasticity of this time period.
In summary, unlike in the mature brain, where chronic alcohol use disorders are characterized by a continuum of graded brain dysmorphology, our preliminary findings suggest that the adolescent hippocampus may be particularly susceptible to the effects of alcohol. The etiology, neuropsychological consequences, and permanence of these changes need to be examined. Future MRI studies of individuals with an adolescent-onset alcohol use disorder as well as of at-risk adolescents are warranted.